
再びバイオ系の学習に戻ります。
前回取りあげた特許では、FAD-GDHという酵素をグルコースセンサに用いていました。
FAD-GDHはヒトの体内には存在しない酵素であり、アスペルギルス属(Aspergillus)と呼ばれるカビの一種などの微生物から抽出した遺伝子を複製して人工的に合成しています。
このようなセンサに使用する酵素は、実験段階、その後の商品化プロセスにおいて大量に必要になりますが、そのような酵素をどのようにして合成しているのでしょうか?
実は、私たちの細胞の中でも日々行われているDNAの複製・転写・翻訳の原理を応用して、これらの酵素は人工的に合成されています。つまり、生体内のしくみを模倣しながら、必要な遺伝子情報を取り出し、それをもとに酵素など目的のタンパク質をつくり出しているのです。
そこで、FAD-GDHをはじめとする「遺伝子から合成される酵素」がどのようなステップを経て作られるのかを、生体内のプロセスとの対比を通じて、3回に分けて整理していく予定です。
第1回目となる今回は「DNA複製」とそれを人工的に行う「PCR」について書きます。
DNAの構造と複製の準備
すべての遺伝情報は、DNAの中に記録されています。まずはDNAの構造と、どのようにしてこの分子が遺伝情報を保持しているのかを見ていきましょう。
DNAの基本構造
遺伝情報を記録する役割を持つ分子がDNA(デオキシリボ核酸)です。
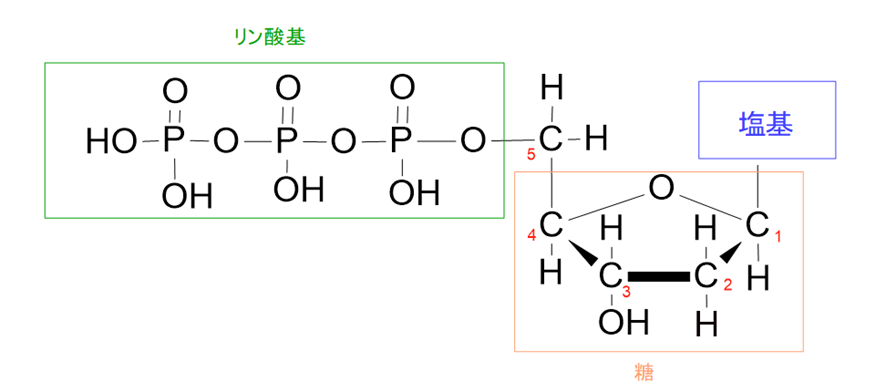
DNAは、糖(デオキシリボース)・塩基・リン酸基の3つの部分で構成された、ヌクレオチドと呼ばれるユニットが長く連なってできています。
糖は5つの炭素からなり、酸素の右隣りに位置する炭素から時計回りに数えて、1番目の炭素に塩基が、5番目の炭素にリン酸基が結合しています。
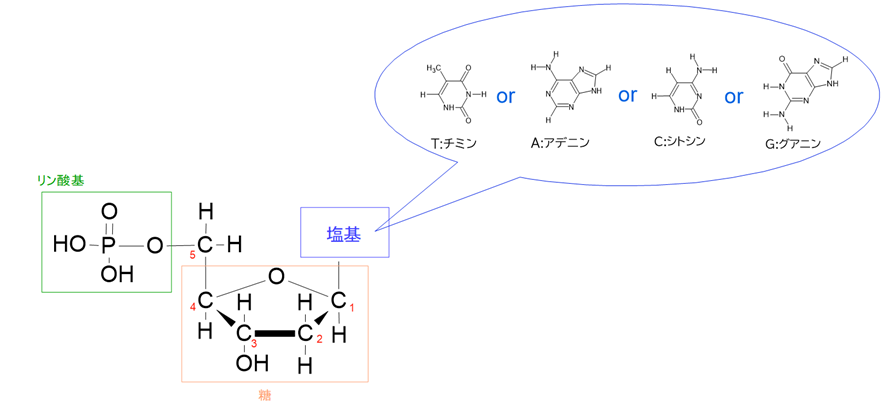
このヌクレオチドのリン酸基の部分が、別のヌクレオチドの3番目の炭素のOH基の部分に結合することで、1本の長い鎖状の高分子ができます。これをDNAと呼んでいます。
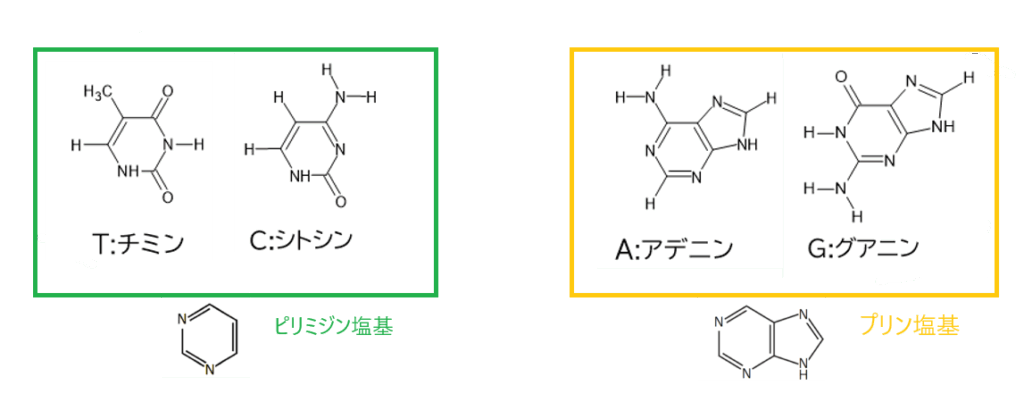
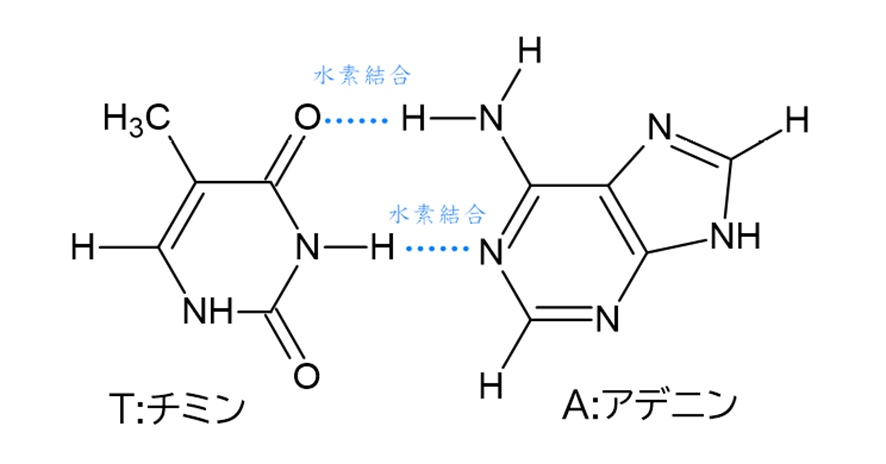
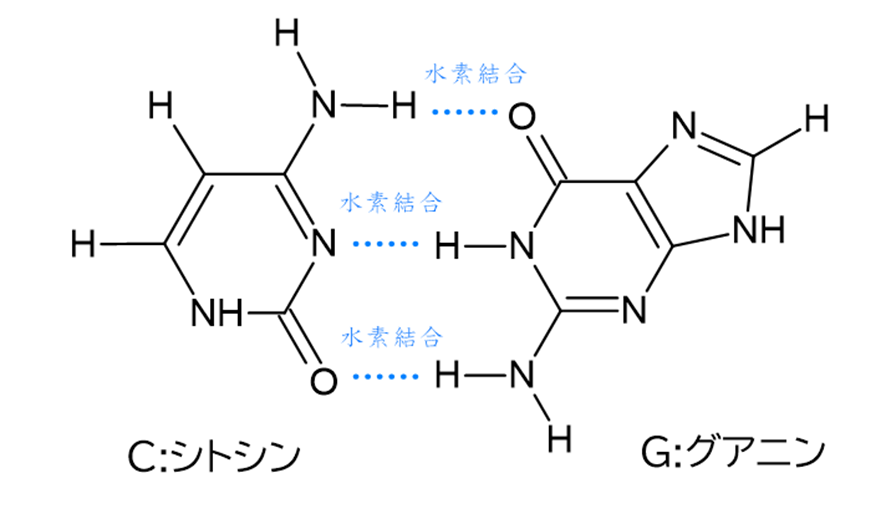
そして、DNAのヌクレオチドの塩基には、A(アデニン)、T(チミン)、G(グアニン)、C(シトシン)という4つの種類があります。
4種類の塩基は大きくプリン塩基とピリミジン塩基に分けられ、
AとGはプリン塩基、CとTはピリミジン塩基に分類されます。
五角形と六角形が合体したような骨格をもつのがプリン塩基で、六角形の骨格をもつのがピリミジン塩基です。
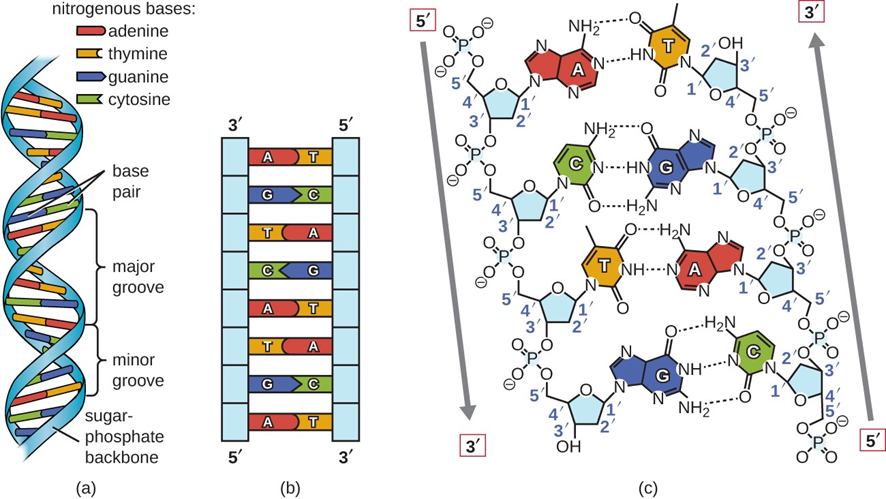
DNAは2本の鎖が絡み合った二重らせん形状のイメージがあるかと思いますが、らせん階段のちょうど段差にあたる部分は、これらの塩基の水素結合により形成されています。
塩基同士の水素結合はどの塩基でも形成されるわけではなく、ちょうどパズルのピースのように、対応する塩基が決まっています。
このような関係を相補的な関係と呼び、ペアになる1組の塩基を塩基対と呼びます。
塩基対を形成するのはAとT、GとCのペアです。ちょうどプリン塩基とピリミジン塩基がペアを作るようになっており、一定の幅の段を持つ階段ができるようになっています。
プリン塩基とピリミジン塩基のペアであれば入れ替えてAとC、GとTでもいいのでは?と思うかもしれませんが、実は入れ替えてしまうと各水素結合の位置が合わず、不安定ならせんになってしまいます。そのため、必ずAとT、GとCという決まった塩基対である必要があります。
クロマチンと染色体の構造
ヒトなどの真核生物のDNAは、タンパク質と組み合わされて普段はクロマチンと呼ばれる状態で核内に存在しています。
このクロマチンは、細胞分裂期になると高度に凝縮され、染色体として観察可能な構造になります。
クロマチンの構造について、以下で説明していきます。
ヒストン、ヌクレオソーム
クロマチンは、DNAがヒストンと呼ばれるタンパク質に巻き付いて形成された糸状の構造です。
通常の細胞活動中(間期)では、DNAはこのクロマチンという糸状の構造で存在しています。染色体は、細胞分裂直前にこのクロマチンが強く凝縮したもので、常に見えるわけではありません。真核生物の染色体は高度に折りたたまれた構造になっています。
まず、二重らせん構造のDNAが、ヒストンというタンパク質に巻き付き、ヌクレオソームと呼ばれる構造を作ります。
ヒストンは、アミノ基(-NH2)を複数持ち塩基性に傾いている塩基性アミノ酸というアミノ酸を多く含んでいます。アミノ基は溶液中では-NH3+となり正電荷を帯びるため、ヒストンは全体として正に帯電しています。このため、リン酸基により負に荷電したDNAと相互作用を生じます。
ヒストンには5つの種類があり、そのうちのH2A、H2B、H3、H4というの4種類のヒストンがそれぞれ2分子ずつ集まって八量体のコアヒストンが構成され、このコアヒストンにDNAが巻き付いてヌクレオソームが形成されます。
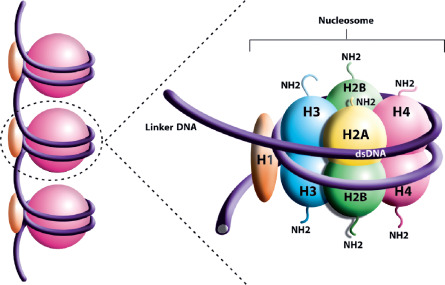
さらに、残りの1種類であるヒストンH1(リンカーヒストン)が、隣接するヌクレオソームの間に結合し、DNAの折りたたみを促進することで、より高次のクロマチン構造(直径約30nmの繊維)を形成するのに寄与しています。
クロマチン繊維は、細胞分裂時にさらに規則正しく折りたたまれ、短く太い染色体へと凝縮されます。これにより、DNAの分配が正確に行われます。
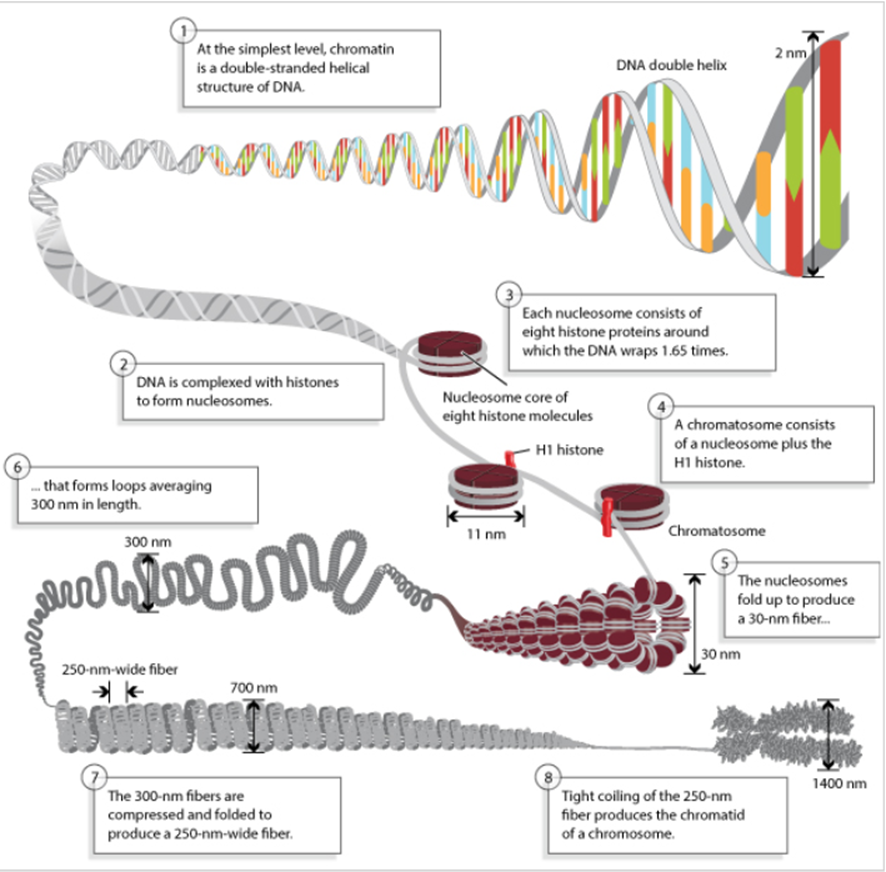
なお、これらの構造はすべて細胞核の内部で形成されており、真核生物におけるDNAは核膜によって囲まれた領域に格納されています。
真核生物は原核生物に比べ複雑に分化した細胞機能を持つため、多くの遺伝情報を必要とします。そのため多くの塩基対(A-TまたはG-Cの塩基のペア)からなる長いDNAを高度に折りたたまれてできる染色体が複数本核内に収容されます。
真核生物の染色体は、基本的には同じ種類の染色体が2本で1組となる二倍体になっています。
エピジェネティック制御
少し補足しておくと、クロマチンは常に上述のように凝縮しているわけではありません。
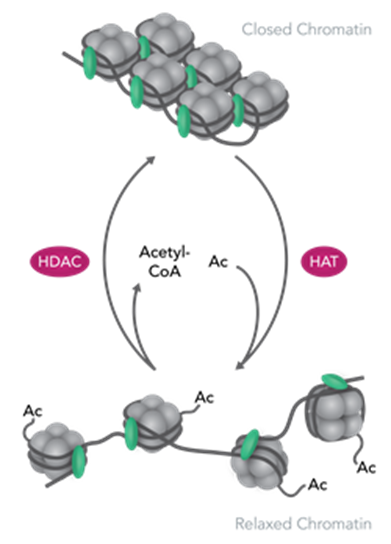
たとえば、DNAへのメチル基の付加(DNAメチル化)、ヒストンへのアセチル基の付加(ヒストンアセチル化)といった化学修飾によって、クロマチン構造がゆるんだり、凝縮したりします。これにより、遺伝子発現のオン・オフが制御されています。
遺伝子発現とはDNAの遺伝情報をもとにタンパク質が作られることをいいます。この点は次回の記事で詳しく取りあげる予定です。
このように、DNAの塩基配列自体は変化させずに、DNAやその周囲にあるヒストンなどのタンパク質に化学的な修飾を加えて、遺伝子の発現を制御するメカニズムをエピジェネティック制御といいます。
以下でクロマチンの凝縮にかかわるDNAメチル化と、クロマチンの開放にかかわるヒストンアセチル化について説明します。
DNAメチル化
4種類の塩基の配列のうち、Cの次にGが続く配列のCにメチル基(-CH3)が結合して5メチルシトシンになることをDNAメチル化といいます。
Cにメチル基(-CH₃)がつくと、そのDNA部分に特定のタンパク質(例:HP1)が結合しやすくなります。HP1は、水に溶けにくい疎水性の部分をもっていて、同じように疎水性部分をもつヒストンと結びつきます。さらに、HP1同士も疎水性どうしで集まりやすいため、DNAがヒストンとともにぎゅっと集まり、クロマチン構造が凝縮します。このような「水になじみにくい部分が集まる性質」を疎水性相互作用と呼びます。
DNAの転写・複製が始まるまではこのような閉じたクロマチン構造が形成されています。
ヒストンアセチル化
一方、後述するDNAの転写が開始する際にはクロマチン構造が開かれます。その際に行われるのがヒストンのアセチル基による修飾、すなわちヒストンアセチル化です。
先ほど、ヌクレオソームの形成に関して、DNAのリン酸基による負電荷とヒストンのアミノ基による正電荷が相互作用することでDNAとヒストンが相互作用すると書きました。
ところが、アセチル基がヒストンのもつアミノ基に結合すると正電荷が弱まり、DNAとの相互作用が弱まり、結果的にヒストンからDNAがほどけやすくなります。
真核生物と原核生物の複製タイミングと構造の違い
染色体は、ヒトのような真核生物と、細菌などの原核生物とでは、異なる構造を持っています。
真核生物と原核生物では、DNAの構造や染色体の形成、複製のタイミングが大きく異なります。
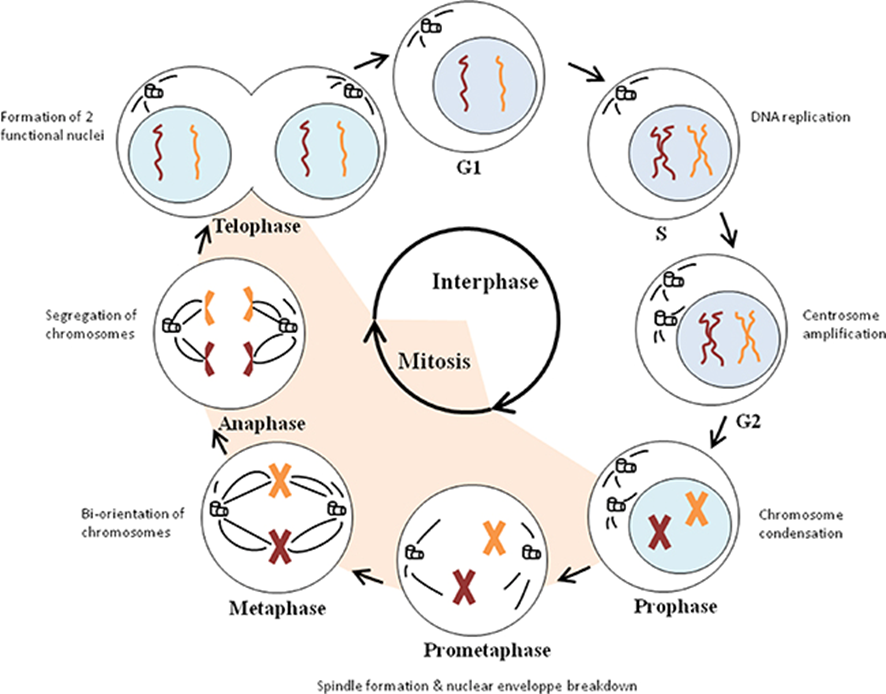
特に真核生物では、上述のようにDNAがヒストンと結びついてクロマチン構造をとり、細胞分裂時には高度に凝縮されて染色体となります。このような複製や凝縮のタイミングは、「細胞周期」と呼ばれる過程で厳密に制御されています。
真核生物の場合
真核生物の細胞は、「細胞周期」と呼ばれる段階を経て、DNAの複製や細胞分裂を行います。細胞周期は大きく分けて以下の4つの時期から構成されています。
- G1期:
細胞分裂を終えた直後の時期で、細胞は大きく成長し、代謝活動が活発になります。この時期、DNAはヒストンと結合して「クロマチン」として核内に広がっており、転写などの遺伝子発現が行われています。 - S期:
DNAの複製が行われる時期です。このときもDNAはクロマチンの状態で存在しており、鋳型として複製されることで、すべての染色体に対応するコピーが作られます。複製後には、それぞれの染色体が2本の姉妹染色分体からなる構造になります。 - G2期:
DNAの複製が完了した後、細胞分裂に向けた準備が進む期間です。タンパク質の合成や細胞小器官の再編成が行われ、この時期もDNAはまだクロマチン状態にあります。 - M期:
核の分裂(有糸分裂)と細胞質の分裂(細胞質分裂)が進行する時期です。このとき初めて、クロマチンが高度に凝縮されて「染色体」として可視化できる形になります。分裂の完了後には、再びクロマチンの状態に戻ります。
DNAは、普段はクロマチンとして広がった状態ですが、M期に入ると初めて染色体として見えるように高度に折りたたまれます。一方、DNAの複製はS期に限って行われており、全体の流れは細胞周期として厳密にコントロールされています。
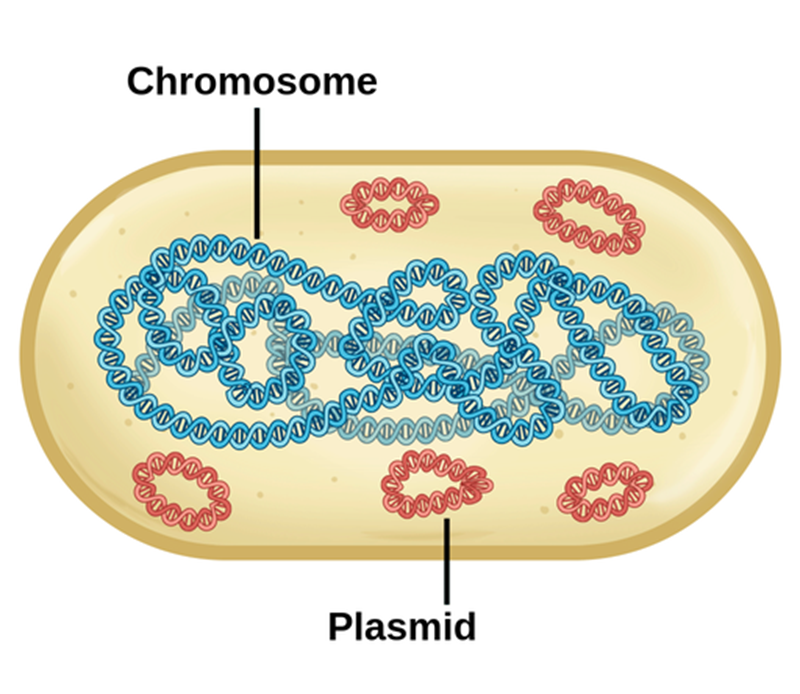
原核生物の場合
原核生物(例:大腸菌)は、細胞分裂とDNA複製が連続的に進むため、真核生物のような細胞周期(G1期→S期→G2期→M期)という明確な段階は存在しません。
成長と同時にDNA複製が始まり、複製が終わると細胞質を分けて分裂します(これを二分裂と呼びます)。
原核生物は、真核生物のような有性生殖ではなく、自らのDNAを複製し、細胞を二分裂することで増殖します。
原核生物の場合は真核生物とは異なり、通常1本の染色体しか持ちません(一倍体)。
また、真核生物はヒストンの結合により直線状のDNAが高度に折りたたまれて複数の染色体を構成していましたが、原核生物の場合は1本のDNAが環状になっています。
真核生物の場合はDNAが直線状のため、テロメアと呼ばれる末端部分が存在します。
テロメアは細胞分裂できるリミットを管理している部分です。テロメアは分裂のたびに少しずつ短くなっていき、ある程度まで短くなるとそれ以上分裂できなくなります。これはテロメア問題と呼ばれており、老化や寿命に関係している可能性があると考えられています。
原核生物のDNAは環状構造をとっているため、テロメアが存在せず、真核生物に見られる「テロメアの短縮による分裂限界(テロメア問題)」は発生しません。
そのため、原核生物の細胞は分裂に関して、老化による制約を受けにくいと考えられています。
また、原核生物には、染色体とは別に、プラスミドと呼ばれる小さな環状DNAを持つことがあります。プラスミドには抗生物質耐性などの補助的遺伝子が含まれており、次回以降で説明する人工的な遺伝子導入にも利用されています。
DNA複製のしくみ
DNAや染色体の構造、複製が行われる時期などを理解したところで、DNAの複製はどうやって行われるのかという本題に入ります。
DNA複製は複数の酵素が協調して進む緻密なプロセスです。
以下では、複製の開始、伸長、終了まで、主要な酵素の働きを中心にそのステップを見ていきます。
DNA複製の開始と進行
複製起点
DNA複製が始まる特定の位置を複製起点と呼びます。
DNAは2本鎖がらせん状になっており、複製を行うにはまずこの2本の鎖をほどく必要があります。どこから2本のDNA鎖をほどき始めるかを決めなければいけませんが、そのスタート地点となるのが複製起点です。
原核生物の場合は、1つの環状DNAの中に複製起点は1つしかありませんが、真核生物の場合は、長い線状のDNAの中にいくつかの複製起点が存在して、同時並行で複製が進みます。
複製起点はA-Tの塩基対が多く含まれることが多くなっています。
これは、G-Cの塩基対では水素結合が3か所あるのに対し、A-Tの塩基対の水素結合は2か所であり、より結合を切断するのが容易であるためだと考えられます。
ヘリカーゼが二重らせんをほどく
二重らせんをほどく役割をするのはヘリカーゼという酵素です。複製起点にヘリカーゼという酵素がくっつき、DNAの二重らせんをほどいていきます。
ヘリカーゼは、六量体でリング状の構造をとっており、その穴の真ん中に一本鎖のDNAを取り囲むように結合します。
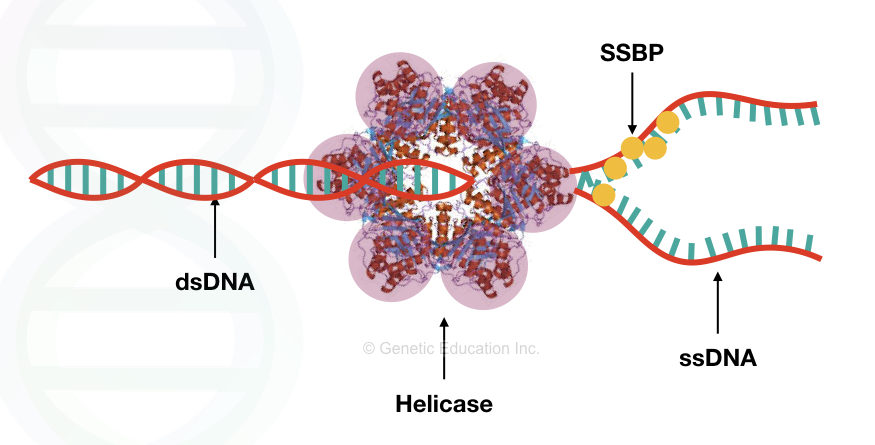
ヘリカーゼはATPやGTPを加水分解することで得られるエネルギーを利用しながら、DNAの塩基対の水素結合をほどいていきます。
結合をほどくということは、現在の状態から別の状態に変化させることになるため、状態を変化させるためのエネルギーを外部から得る必要があります。
そこで、体内でのエネルギー通貨の役割を果たすATPやGTPが活躍します。
ATPやGTPはリン酸基を3つもっており、加水分解によってこのリン酸の結合が切れることで大きなエネルギーが生まれます。
ATP + H₂O → ADP(アデノシン二リン酸) + 無機リン酸 + エネルギー
GTP + H₂O → GDP(グアノシン二リン酸) + 無機リン酸 + エネルギー
ヘリカーゼはATPやGTPから得たエネルギーを使いながら、DNA鎖上を5’→3’方向に進み、DNAの二重らせん構造をほどいていきます。
5’、3’というのは、DNA鎖の向きを表しています。
ヌクレオチドのリン酸基が付いている炭素の位置を5’、水酸基のついている炭素の位置を3′として、それぞれのDNAの末端を5’末端、3’末端、と区別します。
プライム記号(′)は、ヌクレオチド内の糖部分の炭素を指すための記号です。これは、塩基にも炭素番号があるため、それと区別するために使われます。
また、ほどかれたDNA同士が再びくっつかないように、一本鎖結合タンパク質というタンパク質が結合して一本鎖の状態を安定化させています。
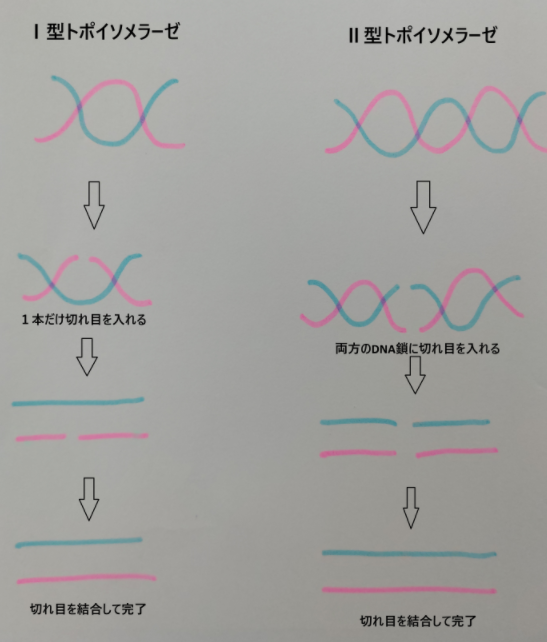
超らせん、トポイソメラーゼ
ヘリカーゼが進んでいくと、その前後は、下図のようにDNAが強く巻かれた超らせんと呼ばれる状態になります。
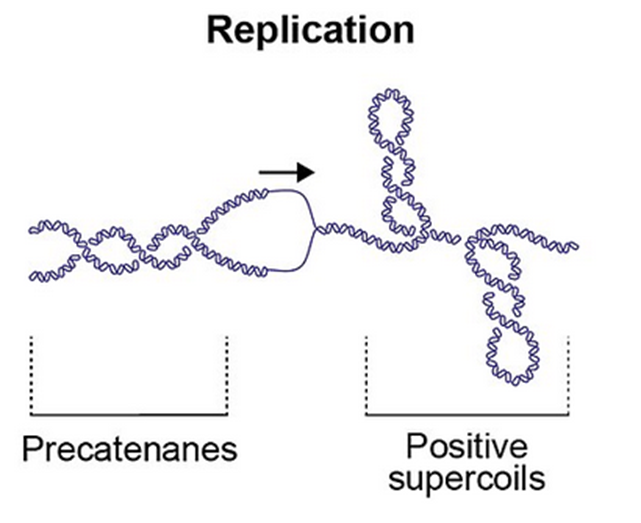
これを解消するのがトポイソメラーゼという酵素です。
ねじれた部分をはさみで切ってねじれを取り除いた後ふたたびつなげ直すような働きをします。
トポイソメラーゼには大きくわけてⅠ型とⅡ型があります。
簡単に言うと、Ⅰ型は、DNAの一本鎖にだけ切れ目を入れてねじれを解消した後、再び結合させます。Ⅱ型は、2本のDNA鎖に切れ目を入れてねじれを解消し、その後再結合させます。
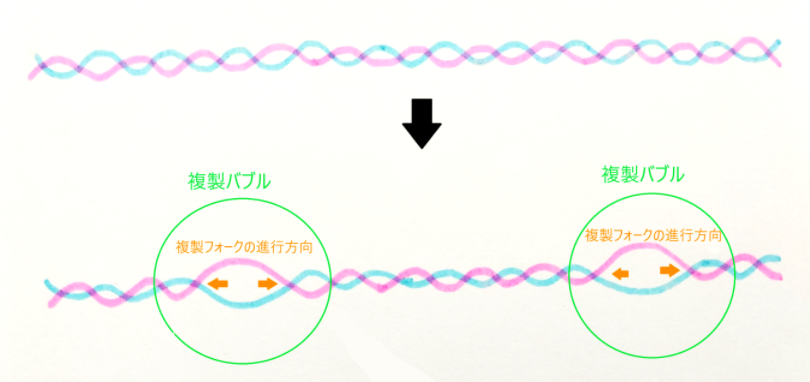
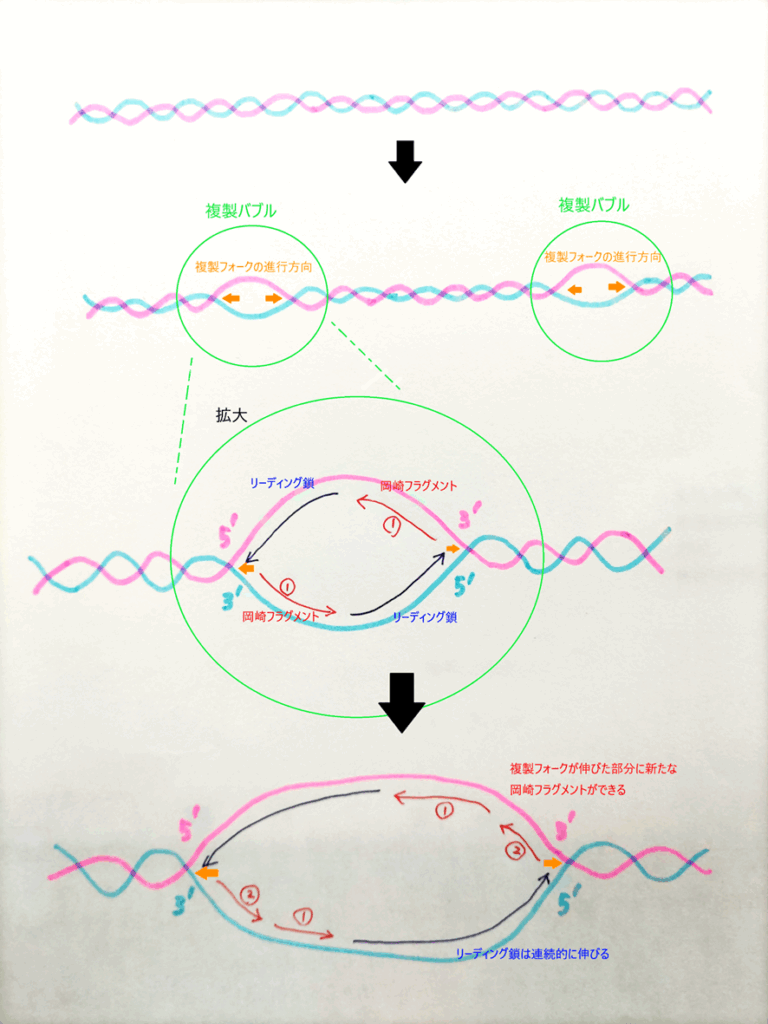
複製フォークの形成
複製が開始する複製起点では、2本のDNAがほどかれることによって、泡のように膨らんだ状態になります。この部分は複製バブルと呼ばれています。
この複製バブルにできるDNAの枝分かれ部分を複製フォークといいます。
ちょうどY字型になっている部分です。先ほど説明したようにDNAヘリカーゼがらせんをほどいていくことにより、例えば下の図であれば、オレンジ色の矢印で示したように、両側に向かってY字の部分が移動していきます。これがフォークの進行方向です。
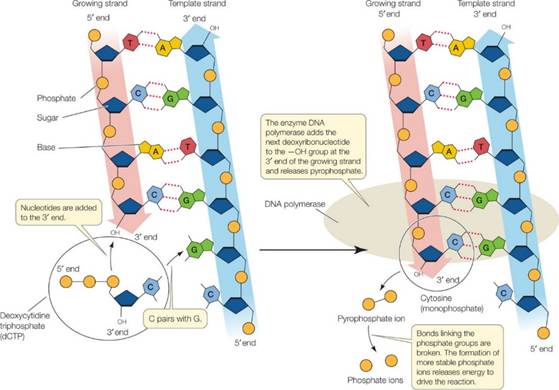
DNAポリメラーゼによる複製
1本にほどけたDNAの塩基は、ペアの相手を失ってしまっています。ここで働くのが、DNAポリメラーゼという酵素です。
DNAポリメラーゼは、細胞の中にあるdNTP(デオキシヌクレオシド三リン酸)というDNAの材料を使って、ぴったり合う塩基を選び、1つずつつなげていくことで、もう1本のDNA鎖(相補的な鎖)を作っていきます。
DNAポリメラーゼは、元のDNA鎖を3′→5′方向に読み取りながら、新しいDNA鎖を5′→3′方向に合成していきます。
つまり、新しく作られるDNA鎖は5′→3′の方向に伸びていきます。
ただし、この「塩基をつなぐ」作業にはエネルギーが必要です。
ここで、dNTPの構造を確認しましょう。
dNTPは、「塩基(A・T・G・C)」「糖(デオキシリボース)」「3つのリン酸基」がセットになった分子です。
DNAとの注目すべき違いは、dNTPでは先ほど触れたATPやGTPと同様にリン酸を3つ持っているという点です。つまり、このリン酸の結合を切ることで大きなエネルギーが出ます。
DNAポリメラーゼはこのリン酸基の結合を切るときに放出されるエネルギーを利用して、次々に塩基をつなげていくのです。
もともと2本のDNAをほどいたので、複製フォークでは1本になったDNAが2本存在し、そのそれぞれが複製されることになります。
実はDNAポリメラーゼは単独で複製を開始することはできず、すでにあるDNAを伸ばしていくことしかできません。そこで、最初にプライマーゼという酵素が短い断片(RNAプライマー)を合成し、DNAポリメラーゼがそのプライマーを足掛かりにしてDNA鎖を伸長させるという流れになります。
リーディング鎖とラギング鎖
ここで、2本のテンプレートとなるDNAは、それぞれ向きが異なるため、複製プロセスが異なります。
リーディング鎖
まずはリーディング鎖から見ていきましょう。
リーディング鎖のもとになる鎖は、フォーク側に5´末端が存在します。
DNAポリメラーゼは、テンプレートとなる鎖を3′→5′方向に読み取ります。この方向はフォークの進行方向と同じなので、DNAポリメラーゼは連続的に新しい鎖(リーディング鎖)を合成することができます。
ラギング鎖
ラギング鎖の場合は、少し複雑になります。というのは、ラギング鎖のもとになるDNAは、フォーク側に3´末端が存在するためです。
先ほど説明した通り、DNAポリメラーゼは3´→5´に向かってテンプレートを読み取って、5´→3´の方向で新しいDNAを合成することしかできません。
ところが、フォークは5′→3′方向に進むため、3′→5′方向に読み取るDNAポリメラーゼとは逆方向になってしまいます。
このため、DNAポリメラーゼはフォークの進行を待って、その都度断片的に新しい鎖を合成することしかできません。このようにしてできる断片的なDNA鎖を岡崎フラグメントといいます。
その後、複数の岡崎フラグメント同士の間を埋めるようにして鎖をつなぎます。
複製の起点にはRNAプライマーが存在するため、複数の岡崎フラグメントの各プライマーを除去し、再びDNAポリメラーゼがそこからDNAを伸長していきます。そして最後にフラグメント同士の間をDNAリガーゼという酵素が結合させることで、全体で1本のラギング鎖ができます。
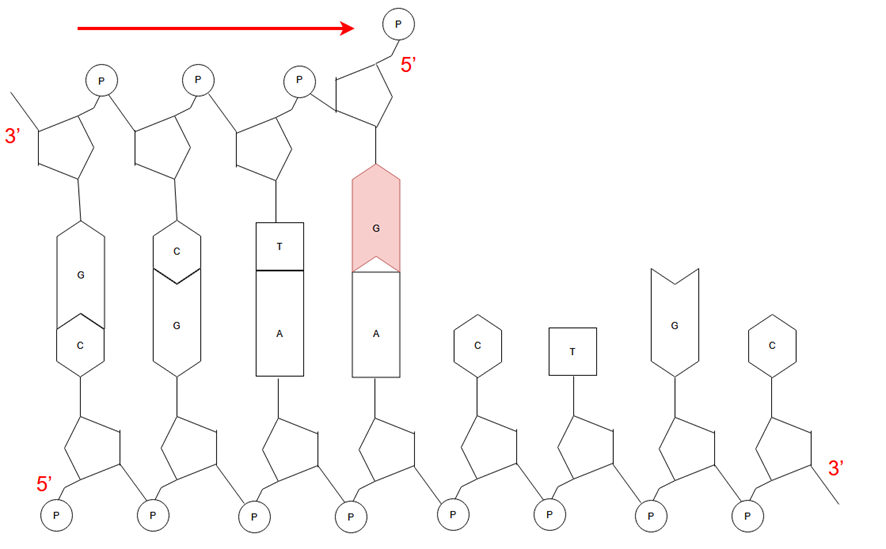
DNA複製方向の制限と理由
ラギング鎖の伸長プロセスを見ていくと非常に複雑に見えます。
DNAポリメラーゼ3´→5´にもDNAを伸長できればこのような複雑さは回避できるのでは?と思うかもしれません。
しかし、DNAポリメラーゼが5´→3´の方向にしか新たなDNAを合成できないのには、理由があります。
DNAポリメラーゼには、つなげるヌクレオチドを間違えた場合にそれを修復する校正という機能を備えており、その校正を可能にするために5´→3´の方向に伸長する必要があるのです。
DNAポリメラーゼは以下のようにDNAを伸長していきますが、途中で間違った塩基を取込むことがあります。
DNAポリメラーゼは、10万塩基に1回という比較的高い確率でミスをしますが、校正機能により、ミスは1000万塩基に1回まで減らすことができます。
DNAポリメラーゼは、ミスを発見すると、3’→5’エキソヌクレアーゼ活性により誤って挿入したヌクレオチドを取り除き、新たに正しいdNTPを取り込んで再びDNA鎖の伸長を続けることができます。
もしもDNAポリメラーゼが3´→5´方向に伸長していたらどうなるでしょうか?
誤りに気付いたDNAポリメラーゼは誤ったヌクレオチドを除去して、正しいヌクレオチドをつなごうとします。
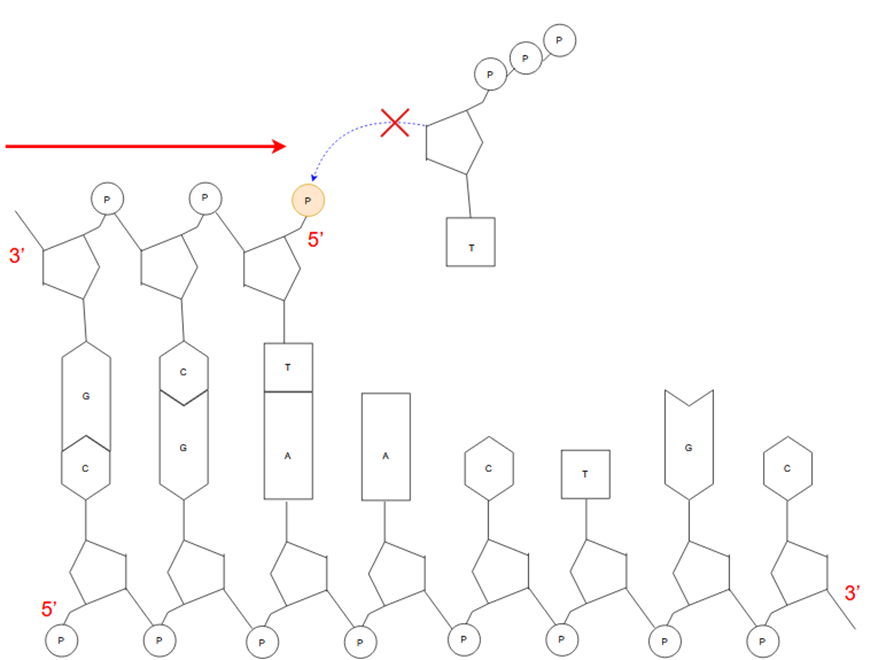
しかし、1つ手前のヌクレオチドのリン酸基は1つしかないため、リン酸基を切断したエネルギーで正しいヌクレオチドをつなぎなおすということができなくなってしまい、そこで伸長がストップしてしまうのです。
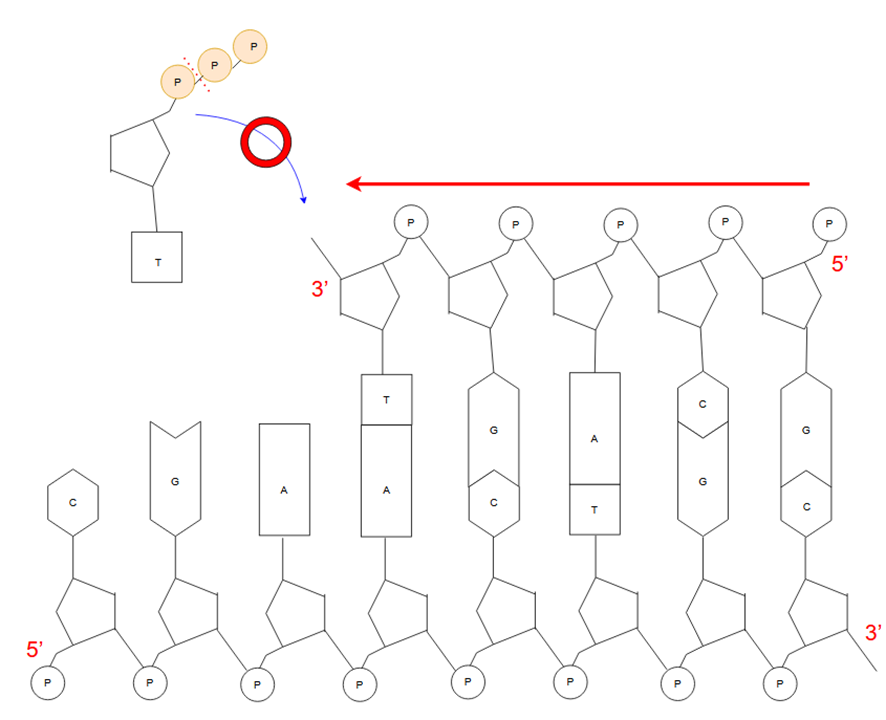
これがDNAポリメラーゼの本来の伸長方向5’→3’の場合であれば、新たにつなぎなおすdNTPの3つのリン酸基の結合を切ったエネルギーで正しいヌクレオチドをつなぎ直し、伸長を続けることができるというわけです。
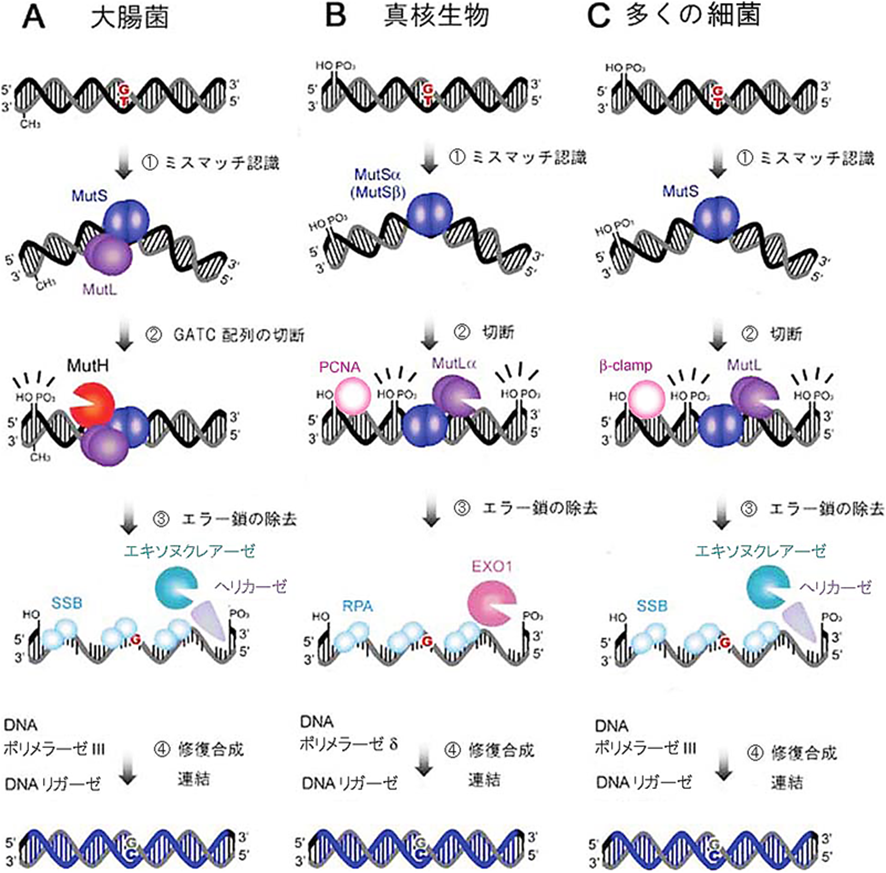
ミスマッチ修復
ちなみに、DNAポリメラーゼによる修復はDNAの複製途中に行われる修復でしたが、DNAの修復は、複製途中以外にも行われています。
例えば、DNAポリメラーゼが気づくことのできなかったミスを、複製完了直後に修復することがあります。これをミスマッチ修復といいます。
例えば、人間も0が連続していると1桁読み間違えるということがよくありますが、似たようなミスで、スリップという現象があります。これは、AAAやTTTの様な同じ塩基が連続する箇所でポリメラーゼが読み飛ばしや余分な塩基の追加を行ってしまうというミスです。
これはポリメラーゼ自身では気づくことができないため、校正が行われないまま複製が完了してしまいます。このほかにも、ポリメラーゼが見落としてそのまま残ってしまったミスが一定数、複製完了時にも存在しています。
それを見つけるのが、原核生物の場合はMutSがMutL、真核生物の場合はMutSαまたはMutSβという物質です。
しかし、すでに複製が完了している状態なので、2本の鎖のうち、どちらが修復すべき対象(新しくできた方の鎖)なのかを見分ける必要があります。
その目印は原核生物と真核生物で異なります。原核生物の場合、複製直後の新生鎖の方の塩基だけ、一時的にメチル基がついていない状態になっており、それを目印にどちらが新生鎖かを見分けます。
真核生物の場合はメチル基による区別は行っておらず、岡崎フラグメントの切れ目など、不連続性がある方を新生鎖として見分けます。
修復すべき部分を見つけたMutSなどは、その修復起点にニックと呼ばれる切れ目を入れます。すると、それを目印にエキソヌクレアーゼという酵素が誤ったヌクレオチドを分解して除去します。その後DNAポリメラーゼが正しいヌクレオチドを合成して、最後にDNAリガーゼが継ぎ目をつなぐことでミスマッチ修復が完了します。
PCR技術―DNA複製の人為プロセス
PCR法(Polymerase Chain Reaction:ポリメラーゼ連鎖反応)とは、目的のDNAを人工的に何百万倍にも増やす技術です。生体内で行われているDNA複製と似た仕組みを、試験管の中で人の手で再現します。
PCRを始めるためには、まず鋳型となるDNAを検体から取り出しておく必要があります。細胞や唾液、血液などのサンプルから、加熱や薬品によって細胞膜を破壊し、中にあるDNAを抽出・精製します。このようにして得られたDNA中に目的の配列が含まれていれば、PCRによってそれを選択的に増幅することが可能になります。
PCRの3ステップ
PCRは、大きく分けて以下の3つのステップからなります。
- ①高温でDNAを1本ずつに分ける熱変性
- ②DNAの目印となるプライマーをくっつけるアニーリング
- ③DNAをのばしていく伸長
それぞれの工程を順番に見ていきましょう。
熱変性
まずは複製の元になる鋳型DNAの2本鎖を1本ずつにほどきます。
生体内ではDNAヘリカーゼによって行われていますが、PCR法では熱を加えることで2本鎖をほどきます。具体的には、94~98℃に加熱することで、2本鎖をつないでいる水素結合が切れて1本の鎖になります。
アニーリング
1本鎖になったDNAに、複製の土台となるプライマーをくっつけます。
生体内ではRNAプライマーが使われていましたが、RNAは熱に弱いため、PCR法ではDNAのプライマーを使用します。
PCRでは、DNAの両端に目印をつける必要があるため、2本のプライマー(左右それぞれの端に結合するプライマー)を使います。
温度を50℃~65℃に下げることで、DNAは再びもとの2本鎖に戻ろうとしますが、DNAプライマーを元のDNAよりも多く含まれるようにすることで、優先的にDNAプライマーと結合させることができます。
伸長
DNAプライマーがくっついたら、そこを起点にDNAを伸ばしていきます。ここでも生体内と同様にDNAポリメラーゼが使われますが、PCRで利用されるTaqポリメラーゼには大きな特徴があり、それは熱に強いということです。
温度を68~72℃まで上昇させるとDNAポリメラーゼが活性化して伸長反応が進みます。
以上の3ステップの操作を1回として、これをn回繰り返すと、
1回で2本(21)、2回で4本(22)、3回で8本(23)、・・・・n回で2n本と増えていきます。
通常、上記サイクルは30回程度繰り返されますので、単純計算で230倍、なんと約10億倍に増幅されることになるのです。
電気泳動による確認
PCRを行ったら、きちんと目的のDNAを複製できているのかを確認する必要があります。
その代表的な方法が、アガロースゲル電気泳動です。
DNAをアガロースゲルという純度の高い寒天の中に入れて電気を流すと、短いDNAは早く進み、長いDNAはゆっくり進みます。進んだ距離を測ることで、どれくらいの長さのDNAが作られたかを確認できます。
DNAはリン酸基により全体に負の電荷を帯びているため、スタート地点を陰極、ゴール地点を陽極にすることで、DNAはスタートからゴールに向かって流れていきます。
アガロースゲルは比較的大きな網目構造になっており、長い分子はその網目に引っ掛かりやすいため進みが遅く、短い分子は比較的進みが早くなります。
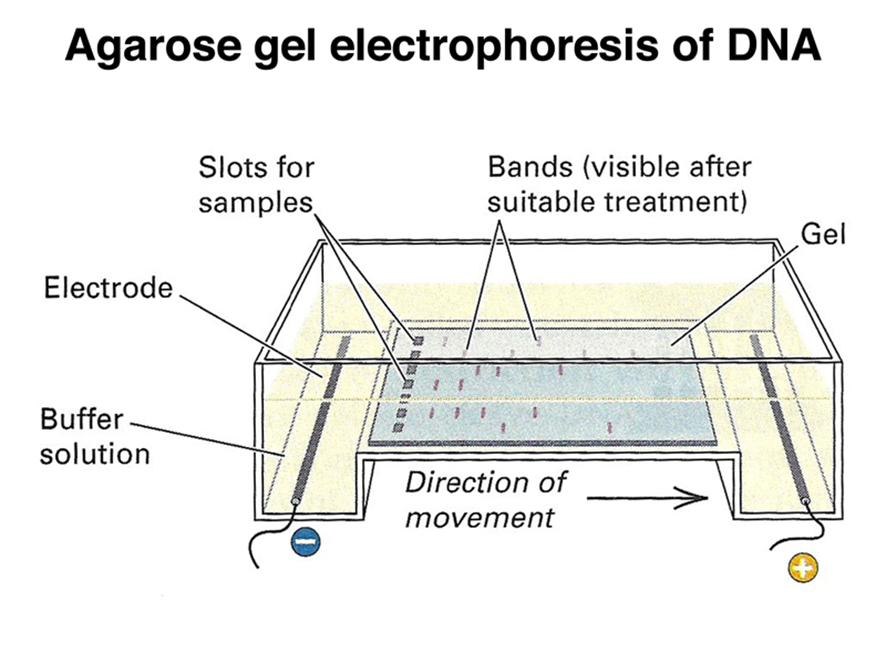
DNAを染色することで、電気泳動後にはバンドが現れます。
バンドの位置は、進んだ距離を表します。
バンドの濃さは、DNAの量を表します。
たくさん作れたDNAほど、バンドが太く・濃くなって見えます。少ししかないDNAなら、バンドは細く・うすくなります。
目的のDNAを一緒に泳動させて比較することにより、欲しいDNAが十分な量合成することができたか確かめることができます。
リアルタイムPCRとその種類
電気泳動の場合は、PCRでの複製が終わった後、目的のDNAが合成できていることを確認できるまで一定の時間を要します。それをリアルタイムで行えるようにしたのがリアルタイムPCRです。
リアルタイムPCRの概要
普通のPCRでは、増えたDNAを後から電気泳動などの別の方法で確認しますが、リアルタイムPCRでは、DNAが増える様子をその場でモニタリングすることができるのです。
PCR試薬の中に蛍光物質を混ぜておき、DNAがきちんと複製されると発光するようにしておくことで、各PCRサイクルで複製状況を確認できるという仕組みです。
リアルタイムPCRにも種類があり、代表的な「SYBR Green法」と「TaqMan法」について以下で説明します。
SYBR Green法
インターカレーター法ともよばれ、SYBR GreenⅠという蛍光物質がよく使われます。
この蛍光物質は、DNAが2本鎖になると、その間に入り込んで光るようになります。したがって、光の変化によってDNAが複製されたことを確認することができます。
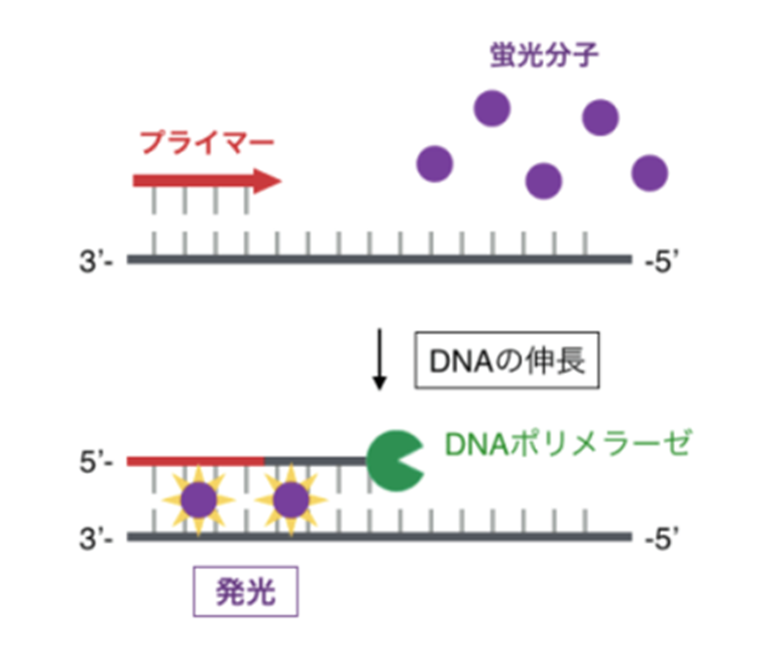
ただし、この方法ではDNAが複製されたということは分かるものの、「どのくらいの長さのDNAができたか」までは直接わかりません。
そこで、増幅されたDNAの融解温度(二本鎖DNAの50%が一本鎖にほどけて遊離する温度)を測って、その値があらかじめ測定しておいた目的のDNAに一致するかどうかを確認し、正しい長さで複製されているかどうかを判断します。
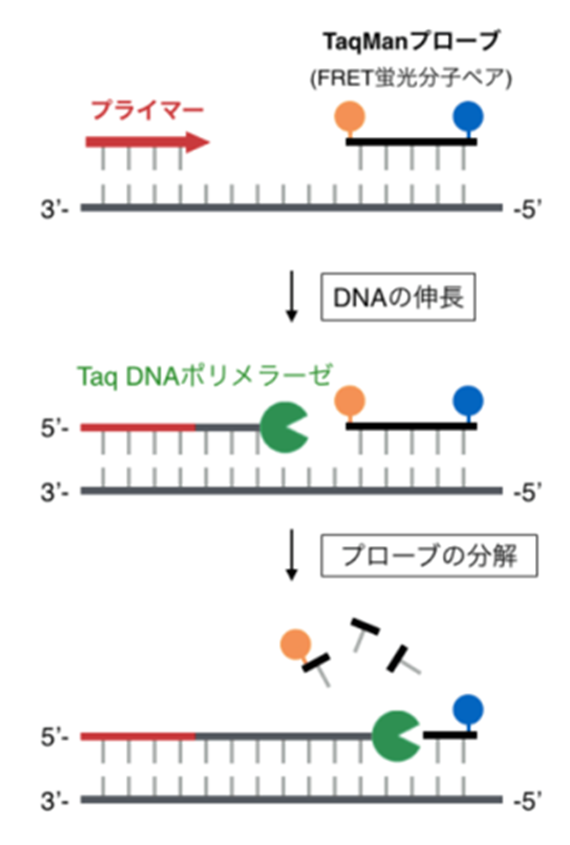
TaqMan法
TaqMan法では、特殊な短いDNA(=TaqManプローブ)を使います。このDNAの両端には、レポータ(光る色素)とクエンチャ(レポータの光を消す色素)がついています。
レポータは単独であれば発光しますが、クエンチャがすぐ近くにあることでFRET(Fluorescence Resonance Energy Transfer)と呼ばれる蛍光エネルギーの移動現象が起きます。つまり、レポータが出す光のエネルギーが、すぐ近くにあるクエンチャに吸い取られてしまい、光らなくなってしまいます。そのため、TaqManプローブは最初の段階では、クエンチャの色は見えていません。
このプローブをプライマーとともに反応系に加えてPCRを行うと、プローブはアニーリングのステップで鋳型のDNAに結合します。
Taqポリメラーゼには、DNAの5′末端側から不要な部分を切るハサミのような機能があり、これを5′→3′エキソヌクレアーゼ活性と呼びます。そのため、5′→3′方向に向かってヌクレオチドを分解することができます。
Taqポリメラーゼは、伸長過程でTaqManプローブのところまで来ると、5′→3′エキソヌクレアーゼ活性によりTaqManプローブを分解します。
プローブが分解されると、レポータとクエンチャが離れるので、レポータが光を出せるようになります。この光を測って、DNAが増えたことがわかるのです。
SYBR Green法は簡便で安価ですが、目的の長さでないDNAにも反応してしまうことがあります。一方、TaqMan法は特定のDNAにだけ反応するので、より正確に調べることができますがより高価になります。
[参考]
・Essential細胞生物学
・タカラバイオ株式会社「PCR 実験の手引き」
・富士フイルム和光純薬株式会社サイト「リアルタイムPCR試薬」






{kind=link}
{kind=link}
コメントを残す