
前々回は生体内でのDNA複製メカニズムとPCR法を、前回はDNAからタンパク質が作られる流れ(セントラルドグマ)について書きました。
では、実際に私たちが必要とするタンパク質を人為的に大量生産するにはどうすればよいのでしょうか?
今回は、グルコースセンサについての記事で取り上げたFAD-GDH(フラビンアデニンジヌクレオチド依存性グルコースデヒドロゲナーゼ)という酵素を大量生産する場合を例にして話を進めたいと思います。
目次
目的遺伝子の準備(cDNA合成)
FAD-GDHは、天然にはカビ(真核生物)で作られるものです。
しかし、カビを直接培養するのは時間とコストがかかり、安定した品質での大量生産は困難です。 そこで、大腸菌を「タンパク質の工場」として利用することで効率的な生産を行うという方法が取られます。
では、カビのFAD-GDHの元になる遺伝子をそのまま大腸菌に入れればFAD-GDH酵素ができるのか、というと実はそう簡単ではありません。
イントロン問題とその解決策
カビ(真核生物)のFAD-GDH遺伝子を大腸菌(原核生物)で発現させる際の大きな課題が、イントロンの処理です。
カビはヒトと同じく真核生物であり、そのDNAには前回説明したように、タンパク質合成には使われないイントロンと呼ばれる配列が含まれます。
元々イントロンを持たない原核生物である大腸菌の中にはこのようなイントロンを除去する機能が備わっていないため、カビのDNAをそのまま大腸菌に入れても目的の酵素をつくることはできないのです。
では、上述の問題を解決するにはどうすればいいのでしょうか?
前回記事で取り上げた真核生物の遺伝子発現の流れを思い返すと、成熟したmRNAであれば、すでにイントロンが除去された状態になっていました。
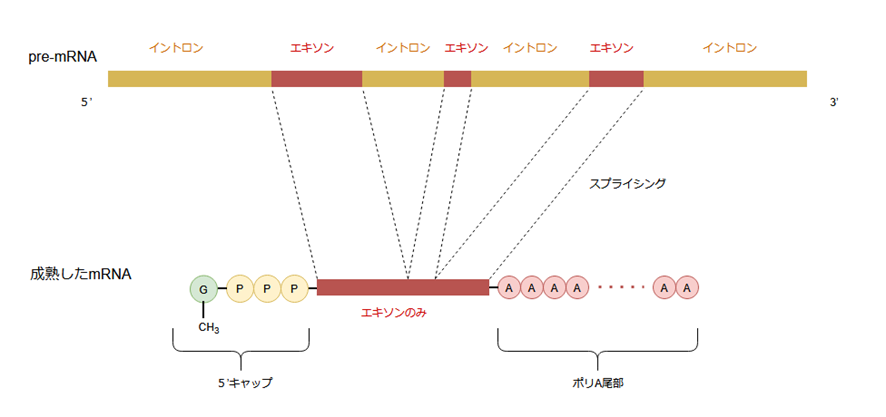
カビの細胞から目的遺伝子のmRNAを取り出し、これを元にDNAを作れば、イントロンが除去された、原核生物でも使えるDNAが手に入ります。
逆転写酵素によるcDNA合成
mRNAからDNAを合成すると言いましたが、前回説明したセントラルドグマに逆行するこのような合成がどうやってできるのでしょうか?
そこで登場するのが逆転写酵素です。
逆転写酵素とは、名前の通り、転写(DNAからRNAを作る)とは「逆」の働きを触媒する、すなわち、RNAからDNAをつくることのできる酵素です。
mRNAを逆転写することにより合成されるDNAは、cDNA(complementary DNA;相補的な DNA)と呼ばれます。
cDNAは以下のような手順で合成されます。
cDNAの合成手順
ステップ1:一本鎖cDNAの合成
前回記事で説明したように、真核生物のmRNAの3’末端にはアデニン(A)が連続して並んだポリA尾部が存在します。
mRNAのこのポリA尾部に、「オリゴdTプライマー」と呼ばれるチミン(T)が連続した短いDNA配列を結合させます。

逆転写酵素は、このプライマーを起点として、mRNAを鋳型にして相補的な一本鎖DNAを合成します。
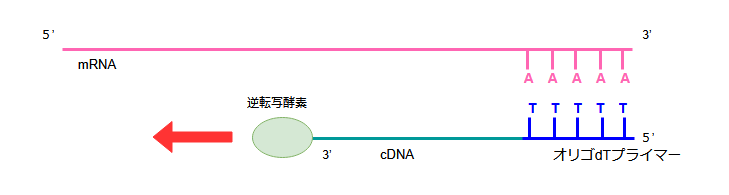
ステップ2:二本鎖cDNAの合成
一本鎖cDNAができた後、元のmRNAをRNase H(RNAを分解する酵素)で除去します。

その後、今度はDNAポリメラーゼを使って、先ほどできた一本鎖cDNAに対して相補的なDNA鎖を合成します。
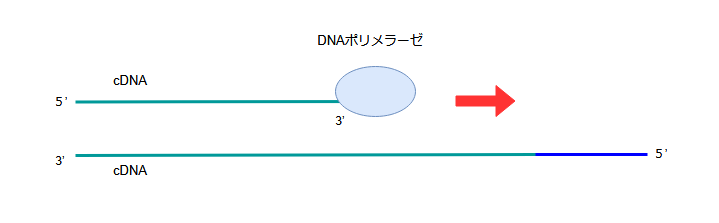
こうして、最終的に二本鎖のcDNAが完成します。

補足:逆転写酵素はなぜ存在する?
非常に都合のよい働きをしてくれる逆転写酵素ですが、本来はRNAを遺伝情報として使うレトロウイルスと呼ばれる種類のウイルスに存在する酵素です。
なじみのある名前としては、ヒト免疫不全ウイルス(HIV)などがレトロウイルスに含まれます。
レトロウイルスは自身のRNAを逆転写酵素で合成したcDNAをヒトなどの遺伝子に挿入することで増殖します。
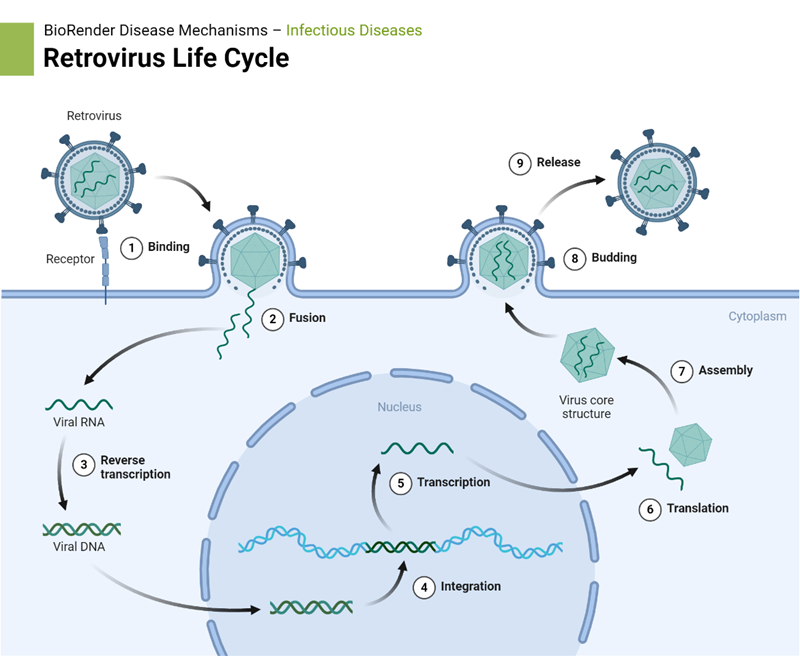
ウイルスと聞くと有害なものというイメージを持つかもしれませんが、その特異なメカニズムをうまく有効活用したのが今回の例というわけです。
発現ベクター
上述の通り、逆転写酵素を利用することでイントロンの問題を解決し、FAD-GDHを合成する設計図となるcDNAが準備できました。
しかし、このcDNAをそのまま大腸菌に導入しようとしても、次のような問題があります。
- 物理的な導入の問題: 遺伝子(DNA)をどうやって大腸菌の中に入れるか?
- 発現制御の問題: いつ、どれくらいの量を作らせるか?
- 遺伝子維持の問題: 細胞分裂しても遺伝子が失われないようにするには?
これらの問題を解決するのが発現ベクターです。
プラスミドベクターの構造
発現ベクターにはいくつかのタイプがありますが、微生物(特に大腸菌)を用いて目的のタンパク質を人工的に合成する場合には、操作性が高く、実績のあるプラスミドベクターが広く利用されています。
プラスミドベクターとは何か
プラスミドとは、天然には原核生物の体内にある、生命維持に必要なDNA情報を含む染色体とは別の、小さな環状のDNAです。
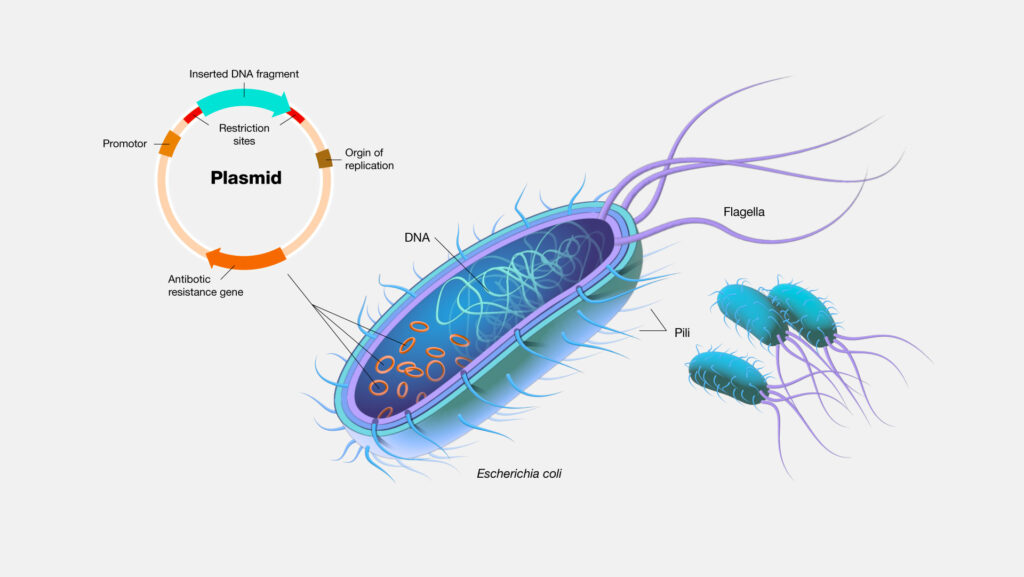
このプラスミドを、発現させたいDNAを埋め込めるよう人工的に改良したものが、プラスミドベクターです。
ベクターの基本要素
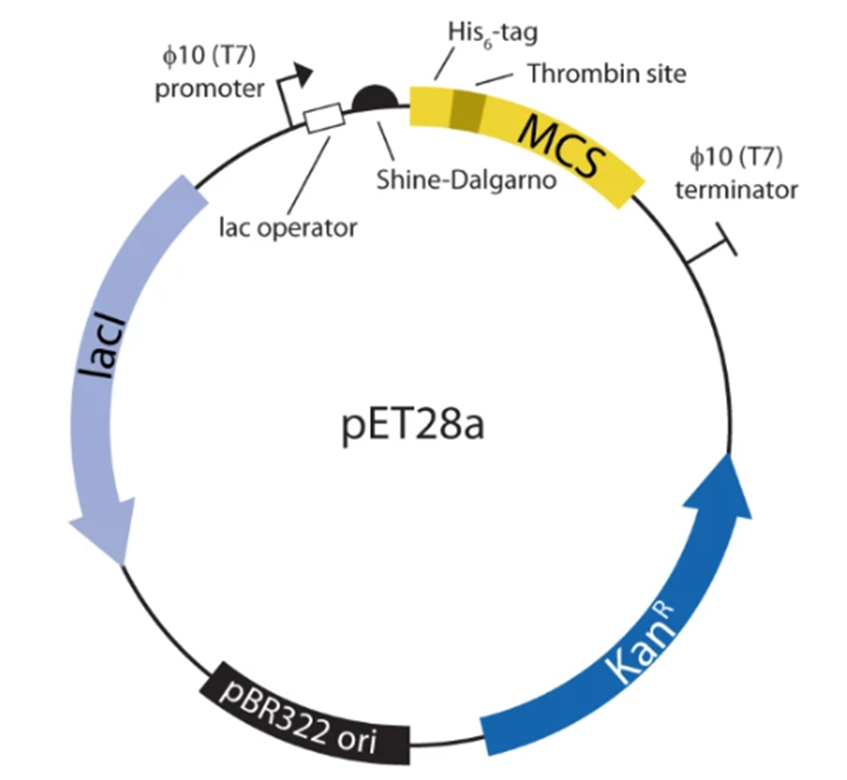
プラスミドベクターは主に以下の基本要素が含まれています。
①誘導性プロモーター (図の「φ10 (T7) promoter」に対応)
②シャイン・ダルガルノ配列 (図の「Shine-Dalgarno」に対応)
③N末端タグ (図の「His6-tag」に対応)
④目的遺伝子領域 (図の「MCS」に対応)
⑤ターミネーター配列 (図の「φ10 (T7) terminator」に対応)
⑥複製起点 (図の「pBR322 ori」に対応)
⑦選択マーカー(図の「KanR」に対応)
それぞれの要素について説明していきます。
誘導性プロモーター
細胞にとって、タンパク質をいつでも大量に作るのは大きな負担です。
そのため、必要なときだけ発現できるように、オペレーターやリプレッサーと組み合わされて「スイッチ」の役割をするのが誘導性プロモーターです。
たとえば、図の「T7プロモーター」は、T7 RNAポリメラーゼのみが特異的に結合して目的遺伝子を発現できます。
T7 RNAポリメラーゼは、大腸菌に自然には存在しませんが、T7ポリメラーゼ遺伝子を染色体に組み込んだ大腸菌株が用いられます。
具体的な制御のしくみについては後述します。
シャイン・ダルガルノ配列
これは前回記事でもふれたように、原核生物に特有の翻訳開始シグナルです。
mRNA中にあるこの配列が、リボソームを構成するrRNAと相補的に結合することで、翻訳の開始位置が決まります。
N末端タグ
発現するタンパク質の先頭(N末端)に目印となる特定の配列をつけることで、タンパク質の検出や精製を簡単にできます。この目印となる配列をタグと呼びます。ちょうど私たちが旅行用のスーツケースに目印となるタグをつけるようなものです。
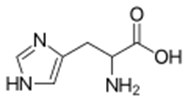
代表的なタグとして、Hisタグを紹介します。
Hisタグは、6~10個程度のヒスチジンというアミノ酸を発現させるタンパク質のN末端に付加したものです。図で「His6-tag」と書かれているのはヒスチジンが6個連なっていることを表しています。
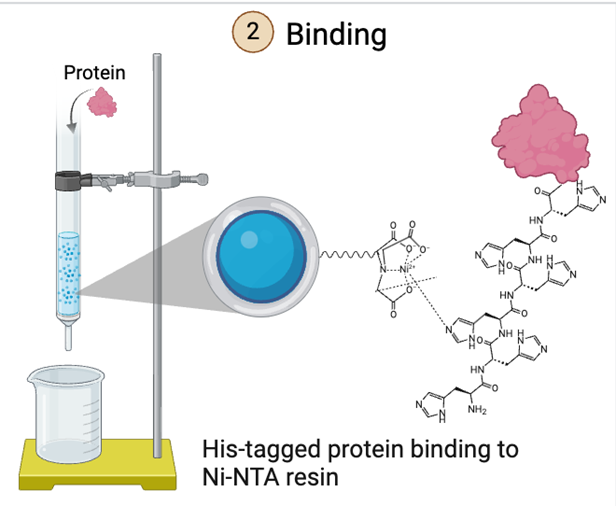
ヒスチジンタグをつけるとなぜ検出や精製が容易になるのでしょうか?
それは、ヒスチジンのイミダゾール環部分がニッケルやコバルトと結合して安定なキレート構造を作るためです。
例えば、ニッケルを含む充填剤を詰めたカラムを使ってクロマトグラフィーを行えば、ヒスチジンタグをつけた目的タンパク質を抽出することができます。
目的遺伝子領域
ベクターの要となるのがこのMCS(multi-cloning site;マルチクローニングサイト)と呼ばれる領域です。
ここには、目的となるタンパク質のDNA、今回の例でいえば、先に準備したcDNAが挿入されます。
先ほど説明したように、cDNAには発現に必要な遺伝情報を含む部分(エキソン)だけが含まれているため、大腸菌でも正確にFAD-GDH酵素を発現できます。
ターミネーター配列
RNAポリメラーゼに転写終了を指示する配列です。
終点を設けることで、目的のmRNAだけを正確に生成することができます。
複製起点
ベクターが宿主細胞の中で自分自身を複製する(DNA複製)ための出発点です。
ここからDNA複製が始まることで、細胞分裂のたびにベクターがコピーされ、ベクターが失われずに維持されます。
選択マーカー
選択マーカーは、ベクターを導入した細胞だけを選び出すための遺伝子です。
例えば、この図のKanRは、カナマイシン耐性遺伝子の略です。
通常の大腸菌はカナマイシンによる抗菌作用で滅ぼされてしまいますが、この遺伝子を持った大腸菌はカナマイシンに対する耐性を獲得します。
そのため、大腸菌の培地にカナマイシンを加えることで、ベクターを持った細胞だけが生き残るようになります。
このように選択マーカーにより、目的の細胞だけを選別・培養することが可能になります。
ベクターへのcDNA挿入
先ほど説明したプラスミドベクターのMCSに、逆転写酵素を使って合成したFAD-GDHのcDNAを組み込む必要があります。
ここで活躍するのが制限酵素です。
制限酵素による切断
制限酵素(restriction enzyme)は、特定のDNA配列を認識して、それを切断する酵素です。
制限酵素の種類によって認識する配列は異なり、また切断後にできる切り口の形にも違いがあります。
片側が他方のDNA鎖より飛び出た形の突出末端になる場合と、両方のDNA鎖が同じように切断される平滑末端になる場合とがあります。
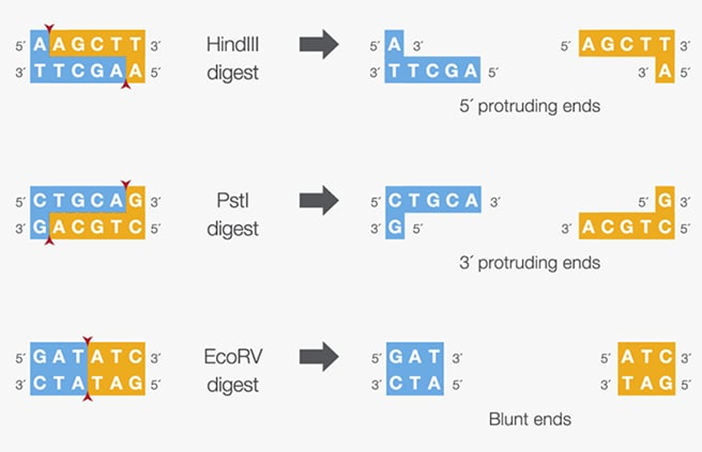
プラスミドベクターには、あらかじめ制限酵素が認識する特定の配列が組み込まれています。
まず制限酵素でプラスミドベクターの特定配列を切断し、挿入したいcDNAの方も同じ制限酵素で切断します。
すると、下図のように、プラスミドベクターの切り口に相補的な末端を持つcDNAをうまく挿入することができます。
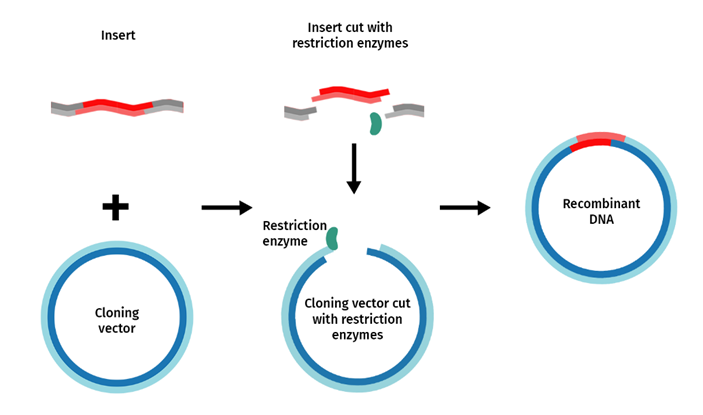
ライゲーション
制限酵素を使って相補的な末端をつくりcDNAを挿入した箇所は、きちんとつなげ直す必要があります。
この役割を果たすのがDNAリガーゼです。DNAリガーゼは、前々回記事で取り上げたDNA複製の際に、岡崎フラグメントをつなぎ合わせる役割でも登場していました。
このDNAリガーゼによって、挿入箇所のDNAの5′ 末端のリン酸基と 3′ 末端の OH 基が結合されて、きちんとつながった環状DNAとなります。
このように、異なるDNA断片同士をつなぐ反応は、ライゲーションと呼ばれています。
宿主の選択
発現ベクターに目的の遺伝子を組み込んだだけでは、まだ目的のタンパク質は得られません。
実際にFAD-GDH酵素を作ってくれる「工場」が必要です。
このような、ベクターの受け取り手となってタンパク質合成を行う生物(細胞)のことを宿主と呼びます。
大腸菌が選ばれる理由
宿主は、目的遺伝子の発現効率、タンパク質の収量や機能性、培養コスト、操作のしやすさなどに直接影響するため、発現系の設計における根本的な選択肢のひとつと言えます。
本稿の対象である酵素(例:グルコースセンサ用酵素など)の人工合成では、以下の理由から原核生物である大腸菌が最も広く使われています。
- 操作が簡単
- 増殖が早い
- 低コスト
大腸菌は、今回説明しているような発現系、遺伝子組み換え、衛生環境のモニタリングなど様々な分野で広く用いられており、操作手順が確立されています。
また、増殖スピードも速く、短時間で大量の細胞が得られます。
大腸菌は、37℃程度の環境で約20分に1回分裂します。そのため、1時間で2の3乗=8倍になり、1日経てば長径2~4 µmほどしかない大腸菌がコップ一杯分の量になるといいます。
培地や設備も簡便で済むので、コストも抑えられます。
形質転換
ここまでで、目的タンパク質のDNAとそのほか必要な要素が組み込まれたプラスミドベクターが完成しました。
次は、このプラスミドベクターを宿主となる大腸菌の細胞内に導入する必要があります。
このように、外部のDNAを取込むことで、元の細胞が取り込まれたDNAの性質に変化することを形質転換といいます。
もともと形質転換とは、一部の微生物が持つ外来DNA取り込み能力による性質変化の現象を指していましたが、現在では人為的なDNA導入技術について多く使われています。
熱ショック法
大腸菌にプラスミドベクターを取込む簡便な方法として、熱ショック法というものがあります。
まず、大腸菌の細胞膜を構成するリン脂質二重層はマイナスの電荷を帯びています。そして、導入したいプラスミドベクターもリン酸基を持っているためマイナスの電荷を持っているため、電荷の反発により細胞膜表面から遠ざけられてしまいます。
そこで、大腸菌を冷やしたCaCl₂溶液と混ぜ合わせると、Caイオンのプラスの電荷により電荷が中和され、細胞膜にプラスミドベクターが近づけるようになります。
さらに、この状態で急激に加熱する(熱ショックを与える)と、大腸菌の細胞膜の流動性が向上し、外来DNAを取込みやすい状態(コンピテントな状態)になります。
また、このとき大腸菌の細胞内外で一時的な温度差が生じます。
一般に、液体の中に温度の高い部分と低い部分があると、全体が均一になるように液体が移動し、対流と呼ばれる流れが生じます。この流れによって、プラスミドベクターの細胞膜への接触や取り込みが促進されます
こうして、プラスミドベクターが効率的に大腸菌の細胞内へ取り込まれます。
エレクトロポレーション法(電気穿孔法)
細胞に高電圧のパルスを加えることによって細胞膜に一時的に孔を開け、そこからプラスミドベクターを導入するという方法です。
高電圧を長時間与えれば細胞膜が破壊されてしまいますが、高電圧をごく短時間のパルスにして印加することで、細胞膜を破壊することなく、孔だけを開けることができます。この孔は一定時間が経つと細胞により修復されます。
急激なpHの変化で細胞を傷つけないように、pHを適正に保つ役割をする緩衝液に大腸菌を懸濁した状態で行います。
熱ショック法と比べて導入効率が高く、美容液成分を肌に効率よく浸透させるために美容分野でも使われている方法ですが、エレクトロポレーション用の専用装置が必要になります。
発現誘導
プラスミドベクターの構造説明のところでも簡単に書きましたが、目的のタンパク質を常に大量生産すると、宿主にとって大きな負担となります。そのため、必要な時だけ発現させる制御機構が重要です。このような制御された発現を開始する操作が発現誘導です。
lacオペロンの基礎
前述のとおり、今回例として挙げているプラスミドベクターには、T7プロモーターというプロモーターが組み込まれています。このようなT7プロモーターを使用する発現ベクターは、pETという名前で呼ばれています。
ここでは、pETベクターにIPTG(イソプロピル-β-D-チオガラクトピラノシド)という物質を組み合わせることで行われる発現誘導、すなわちIPTG誘導を紹介します。
IPTG誘導のしくみに入る前に、まず、大腸菌に元々備わっているラクトースオペロン(lacオペロン)について説明します。
オペロンとは、あるタンパク質の発現にかかわる一連の遺伝子群のことをいいます。
ラクトースオペロンとは、ラクトースを分解してエネルギー源となるグルコースに変えるための働きをする酵素を作り出す一連の遺伝子群のことです。
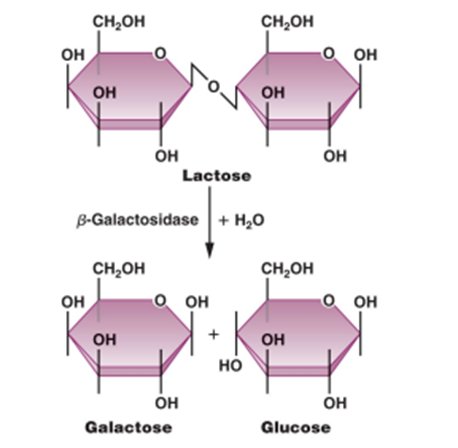
ラクトースが存在するときには分解酵素を作り、存在しない場合には作らない、といういわば省エネシステムです。
lacオペロンは次の要素で構成されています。
- lacリプレッサー
- [lacプロモーター] ─ [lacオペレーター] ─ [lacZ] ─ [lacY] ─ [lacA]
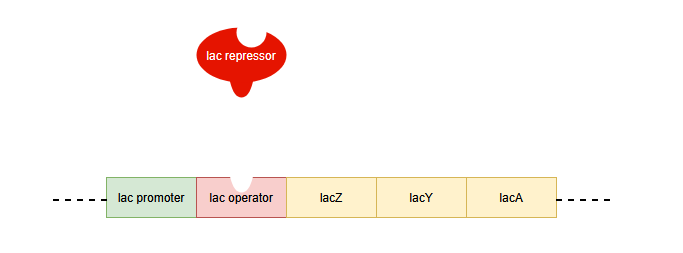
lacZ、lacY、lacAは、それぞれ、ラクトースを分解するのに必要な酵素を作るための遺伝子です。
RNAポリメラーゼがlacZ、lacY、lacAの部分を読んで転写することで、ラクトースの分解に必要な酵素の設計図となるmRNAが作られることになります。
lacプロモーターは、RNAポリメラーゼが転写を開始するスタート位置です。
そして、lacプロモーターとlacZ、Y、Aとの間にはlacオペレーターが存在します。この部分にlacリプレッサーが付いたり離れたりすることで、RNAポリメラーゼの動きを制御します。
以下で図を参照しながら確認しましょう。
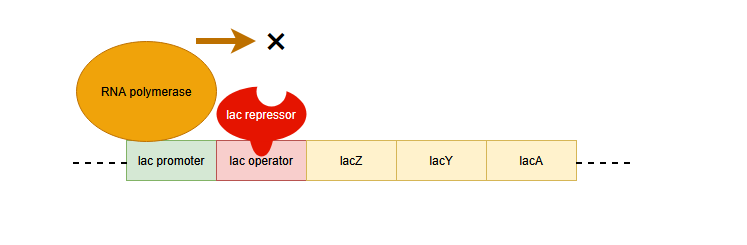
ラクトースが存在しない場合
ラクトースが存在しないとき、lacリプレッサーがlacオペレーターに結合します。
すると、RNAポリメラーゼはlacZ、Y、Aの転写を行うことができないため、mRNAが合成できず、目的の酵素が作られません。
ラクトースが存在する場合
ラクトースが存在すると、ラクトースがlacリプレッサーに結合し、lacリプレッサーの構造を変化させます。すると、lacリプレッサーはlacオペレーターに結合しなくなり、RNAポリメラーゼはlacZ、Y、Aを読んでmRNAを作ることができます。このmRNAが翻訳されて目的の酵素が作られます。
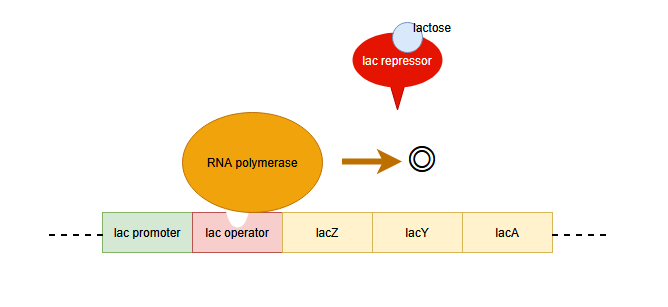
このような抑制因子による転写制御は、「負の制御」または「負の調整」と呼ばれます。
カタボライト抑制
話を簡単にするためにラクトースの有無のみを取り上げましたが、実際の大腸菌ではラクトースの有無だけでなく、グルコースの有無も重要です。
なぜグルコースが重要なのでしょうか?
グルコースは最も効率的に利用できる糖源です。そのため、グルコースとラクトースが同時に存在する場合は、グルコースを優先的に利用し、ラクトースの分解酵素はほとんど作りません。このような現象をカタボライト抑制と呼びます。
カタボライト抑制のメカニズム
カタボライト抑制には、CRP-cAMP複合体という分子が重要な役割を果たしています。CPRは、cAMP受容タンパク質と呼ばれ、cAMPという鍵が特異的に結合できる鍵穴を持つようなタンパク質です。
CRPとcAMPが結合してできた複合体タンパク質は、DNAに結合することで転写を促進する役割を果たします。CRP-cAMPのように遺伝子の転写を促進するタンパク質はアクチベータと呼ばれます。
アクチベータがあることで転写が進むことから、このような転写制御は、「正の制御」あるいは「正の調整」と呼ばれます。
グルコースが存在する場合:
グルコースが存在する場合には、大腸菌細胞内のcAMP濃度が低下します。cAMPが少なければ、CRPとcAMPの複合体の形成も抑制されます。この複合体がないと、lacプロモーターでの転写が効率的に進まないため、ラクトース分解酵素の産生が抑制されます。
グルコースが存在しない場合:
cAMP濃度が上昇することで、CRP-cAMP複合体が形成されます。この複合体がlacプロモーター領域に結合することで、RNAポリメラーゼの転写開始が促進され、ラクトース分解酵素が効率的に作られます。
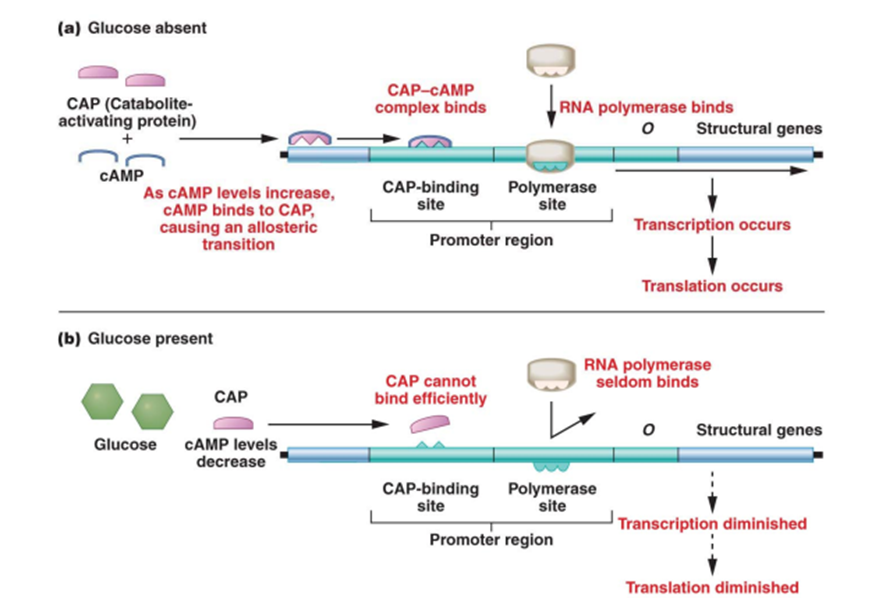
つまり、ラクトースを分解する酵素が大量に作られるのは、「ラクトースがあり、かつグルコースがない」場合のみです。
効率の良いグルコースから先に使うことで、無駄な酵素産生を防ぎ、エネルギー効率を最大化した戦略をとっているのです。
IPTG誘導のしくみ
では、ラクトースオペロンの基本的な仕組みが分かったところでIPTG誘導の話に進みましょう。
大腸菌のもともと持っている染色体の中には、先ほど説明した通りlacオペロンが備わっています。
またさらに、T7プロモーターに特異的に結合するT7 RNAポリメラーゼを発現するための遺伝子を導入済みの大腸菌が使われます。

一方、導入したプラスミドベクターは以下のような構成です。
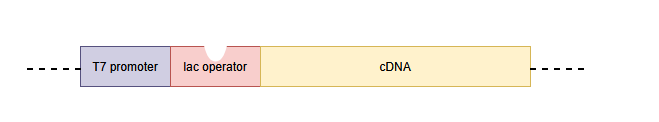
ラクトースを加えると、まずラクトースとlacリプレッサーが結合して、大腸菌の染色体内のlacオペレーター部分とlacリプレッサーの結合が防がれ、ポリメラーゼがT7 RNAポリメラーゼDNAを転写できるようになり、結果として、T7 RNAポリメラーゼが合成されます。
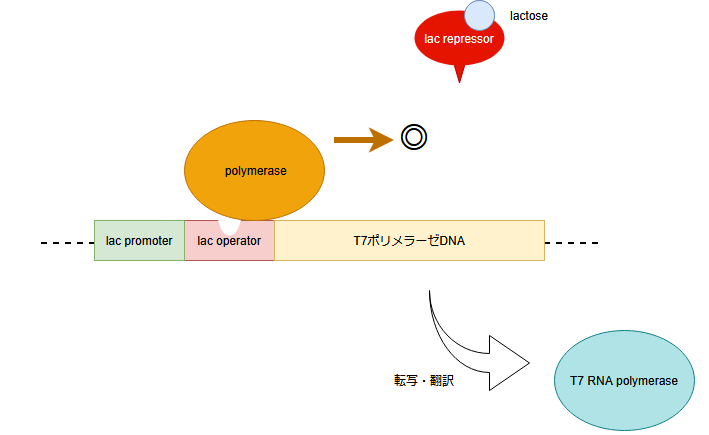
さらに、ラクトースはプラスミドベクターのlacリプレッサーにも結合し、lacリプレッサーがプラスミドベクターのlacオペレーター部分に結合するのを防ぎます。こうすることで、先ほど作られたT7 RNAポリメラーゼがプラスミドベクターのT7プロモーターに結合して、cDNAの転写を開始し、最終的に目的のタンパク質が合成されます。
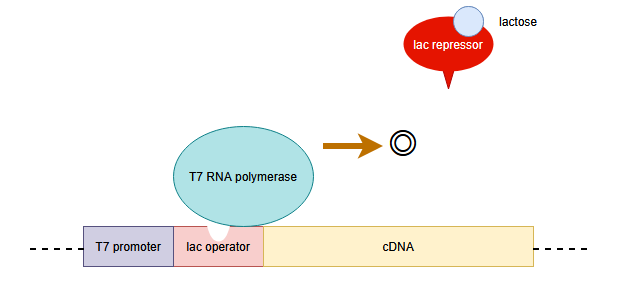
しかし、ここでひとつ問題が出てきます。
cDNAを発現させるためにラクトースを与えると、大腸菌の染色体にもともとあるlacZ、lacY、lacAも転写されて、ラクトースの分解酵素が合成されてしまうのです。ラクトースの分解酵素が合成されれば、与えたラクトースの濃度は減少していき、プラスミドベクターの発現が不安定になってしまいます。
そこで役立つのがIPTGです。
IPTGは、ラクトースと同様にlacリプレッサーに結合してlacリプレッサーがlacオペレーターに結合するのを防ぐことができますが、その一方でラクトースの分解酵素によって分解されることはありません。
したがって、目的のcDNAを発現させたいときにIPTGを与えることで、宿主の大腸菌に大きなダメージを与えることなく、必要なときにだけ目的のタンパク質を合成できるようになるのです。
二重制御システムの理由
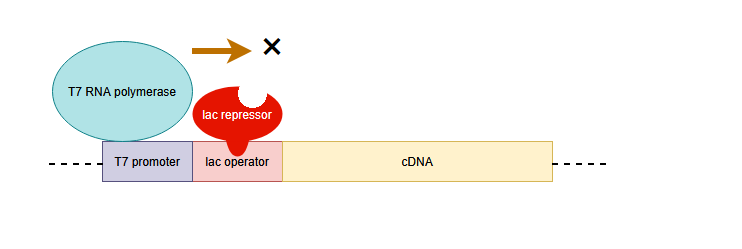
大腸菌側にT7RNAポリメラーゼ遺伝子の発現をコントロールするためのlacオペレーターがすでに入っているのに、なぜ、プラスミドベクター側にもlacオペレーターを入れて二重制御システムにしているのでしょうか?
それは、大腸菌が本来持っているRNAポリメラーゼと比べてT7 RNAポリメラーゼの活性が非常に高いためです。
目的遺伝子の発現をストップさせるためにIPTGが存在しない状態になっても、T7 RNAポリメラーゼが微量でも存在していれば、ベクターのT7プロモーターに結合して転写を開始してしまいます。このように、目的遺伝子の発現をストップさせているはずなのに「漏れ」が生じてしまう現象は、Leaky expressionやBackground expressionと呼ばれています。
プラスミドベクターにもlacオペレーターを入れておけば、T7 RNAポリメラーゼが存在していてもIPTGが存在しなければ目的遺伝子の転写は行われないため、発現をより正確にコントロールできるのです。
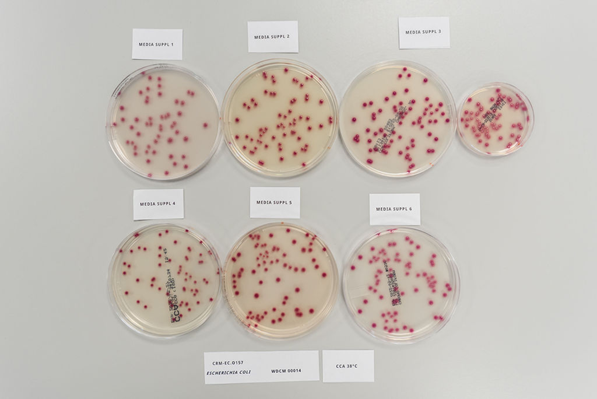
大腸菌の培養
プラスミドベクターを導入した大腸菌は培地と呼ばれる場所で増殖されます。
先ほどのIPTGや、目的の遺伝子を持たない大腸菌を死滅させるためのカナマイシンなどがこの培地の中に含まれます。
前述の通り、大腸菌は人の体温程度の環境でどんどん増殖していき、コロニーと呼ばれる目に見える塊になっていきます。
この各大腸菌の体内で目的のタンパク質が合成されているはず、ということです。
ただし、目的遺伝子が高発現だからといって常に良いとは限らないため、その点は注意が必要です。
タンパク質が多く作られすぎると、凝集して不溶性のインクルージョンボディとなってしまいます。インクルージョンボディになると本来のタンパク質としての機能を発揮することができません。
発現確認-ウェスタンブロッティング
大腸菌を培養したところで、実際に目的のタンパク質が作られていなければ意味がありません。そこで、目的のタンパク質が正しく作られているかを確認する必要があります。
その中でも代表的な方法がウェスタンブロッティングです。
基本的な考え方
1. タンパク質を大きさ別に分ける(SDS-PAGE)
2. 目的のタンパク質だけを見つける(抗体反応)
3. 結果を可視化する(検出)
これはPCR後にアガロースゲル電気泳動でDNAを確認したのと似ていますが、タンパク質用にアレンジした手法です。
前回説明したアガロースゲル電気泳動との違い
- ゲルの種類:ポリアクリルアミドゲル(より細かい網目)
- 対象物質:DNA → タンパク質
- 電荷の調整:SDSでタンパク質をマイナス電荷に調整
ウェスタンブロッティング法の流れをざっくりと説明すると、以下のような流れになります。
①SDS-PAGE
②メンブレンへの転写
③ブロッキング
④1次抗体反応
⑤2次抗体反応
⑥検出
以下でもう少し細かく説明します。
SDS-PAGEによるタンパク質分離
SDS-PAGEは、SDSという界面活性剤を利用したポリアクリルアミドゲル電気泳動です。
ポリアクリルアミドゲルはアガロースゲルに対して網目が小さいため、アガロースゲルに使用するよりも低分子量の物質を分離するのに適しています。
タンパク質は一般的にアガロースゲルで分離するDNA断片よりも分子量が小さいため、より網目の小さなポリアクリルアミドゲルが適しています。
また、DNAの場合はリン酸基によって常にマイナスの電荷を帯びているため、スタートにプラス、ゴールにマイナスの電荷をあたえれば自然とゲルの中を泳動させることができたのですが、タンパク質の場合はアミノ基(溶液中でプラスの電荷を帯びる)とカルボキシル基(溶液中でマイナスの電荷を帯びる)の両方を有するため、それぞれの基の割合によってタンパク質全体の帯びる電荷が変わり、場合によってはうまく泳動しません。
そこで、SDSという陰イオン界面活性剤を添加してタンパク質全体がマイナスの電荷を帯びるようにすることで、安定した泳動を可能にしています。
メンブレン転写
SDS-PAGEによりタンパク質を分子量ごとに分離したら、今度はそれをニトロセルロースまたはポリフッ化ビニリデン(PVDF)でできたメンブレンに転写します。
ポリフッ化ビニリデン(PVDF)の場合は、使用前にメタノールで親水化する必要があります。
ろ紙の間に電気泳動後のゲルとメンブレンを挟み、メンブレン側がプラス極になるようにして電圧を印加すると、電気泳動と同じような要領でゲルのタンパク質がメンブレンに転写されます。

なぜゲルをそのまま使わずにわざわざメンブレンに転写するのかというと、メンブレンの方がゲルよりも強度が高く、その後の抗体を用いるプロセスでゲルでは抗体が十分に浸透しないためです。
抗体反応による検出
- ブロッキング
抗体反応の前にメンブレン上のタンパク質が存在しない空白部分をマスキングして、抗体反応時にその空白部分に抗体が結合してしまうのを防ぐ必要があります。これがブロッキングです。
ブロッキングに使用する試薬には、メンブレンにはよく吸着し、なおかつこの後に使用する抗体とは反応しないタンパク質として、スキムミルクやウシ血清アルブミンが使用されます。
- 1次抗体反応
ターゲットのタンパク質のみと反応する(特異的に反応する)抗体を使用して、抗体を結合させます。
抗体は特定のタンパク質(抗原)にのみ結合する性質があります。
FAD-GDH専用の抗体を使うことで、他の大腸菌タンパク質と区別して検出できます。
- 2次抗体反応
1次抗体反応で使用した抗体に特異的に結合する抗体を使用します。
2次抗体はラベルの役目を果たし、目的のタンパク質を検出しやすくします。
検出方法の比較
2次抗体の標識として、化学発光法と発色法という2種類が存在します。
化学発光法
化学発光法でよく用いられるのは、HRP(ホースラディッシュペルオキシダーゼ)標識という、西洋ワサビから発見された酵素です。
この酵素がルミノールという基質の過酸化水素(H2O2)による酸化反応を触媒します。酸化ルミノールは励起状態になり、基底状態に戻る際に放出するエネルギーで発光します。
この光は非常に微弱なため肉眼ではほとんど見えず、X線フィルムやCCDカメラで確認します。
発色法
HRP標識など二次抗体の標識となる酵素に反応して不溶性の着色物質を形成するような発色基質を使用する方法です。
発色法は化学発光法に比べて検出用の機器を使わずに肉眼で確認できる点で簡便ではありますが、化学発光法の方に比べ検出の感度は劣ります。
精製と活性評価
ウェスタンブロッティングなどで目的のタンパク質が合成されているということが確認できたら、次はそのタンパク質だけを取り出し、その活性を評価するという手順が必要になります。
発現したタンパク質を他の細胞成分から分離し、純度の高い状態で回収することが求められます。これをタンパク質の精製と呼びます。
この際に使われるのが、プラスミドベクターの構成の部分で説明したように、ヒスチジンタグなどを用いて目的のタンパク質だけを抽出します。
続いて、目的のタンパク質の活性を評価します。
いくらタンパク質が多く作られていても、正しく機能してくれるものでなければ意味がないためです。
たとえば、グルコースセンサ用の酵素であれば、以前の記事で説明したように、グルコース量の変化に応じた溶液の色変化を見る(比色法)、酵素反応の際に生じる電子の動きを電流で測る(電気化学測定法)といった方法です。
このように、活性測定によって「機能するタンパク質」が得られたと確認できて、ようやく目的のタンパク質が得られた、ということになるのです。
[参考]・Essential細胞生物学
![[商品価格に関しましては、リンクが作成された時点と現時点で情報が変更されている場合がございます。]](https://hbb.afl.rakuten.co.jp/hgb/4a47823f.ec7d24f1.4a478240.065a96b7/?me_id=1213310&item_id=20399941&pc=https%3A%2F%2Fthumbnail.image.rakuten.co.jp%2F%400_mall%2Fbook%2Fcabinet%2F6825%2F9784524226825_1_2.jpg%3F_ex%3D240x240&s=240x240&t=picttext "[商品価格に関しましては、リンクが作成された時点と現時点で情報が変更されている場合がございます。]")

・Thermo Fisher SCIENTIFIC「ベクターマップの読み方~クローニング・タンパク質発現で迷子にならないために」
・東端 啓貴「大腸菌を宿主とした異種タンパク質高発現のイロハ」(生物工学、第91巻、第2号、2013年)
・MBL株式会社「ポリアクリルアミド電気泳動(SDS-PAGE)の原理と方法」







コメントを残す